
Online RLadies
@SherlockpHolmes
The slides : https://bios221.stanford.edu/NetworkSlides.html
See Github repo: spholmes/GraphsTutorial
Install with:
devtools::install_github("spholmes/GraphsTutorial")Online RLadies
@SherlockpHolmesThe slides : https://bios221.stanford.edu/NetworkSlides.html
See Github repo: spholmes/GraphsTutorial
Install with:
devtools::install_github("spholmes/GraphsTutorial")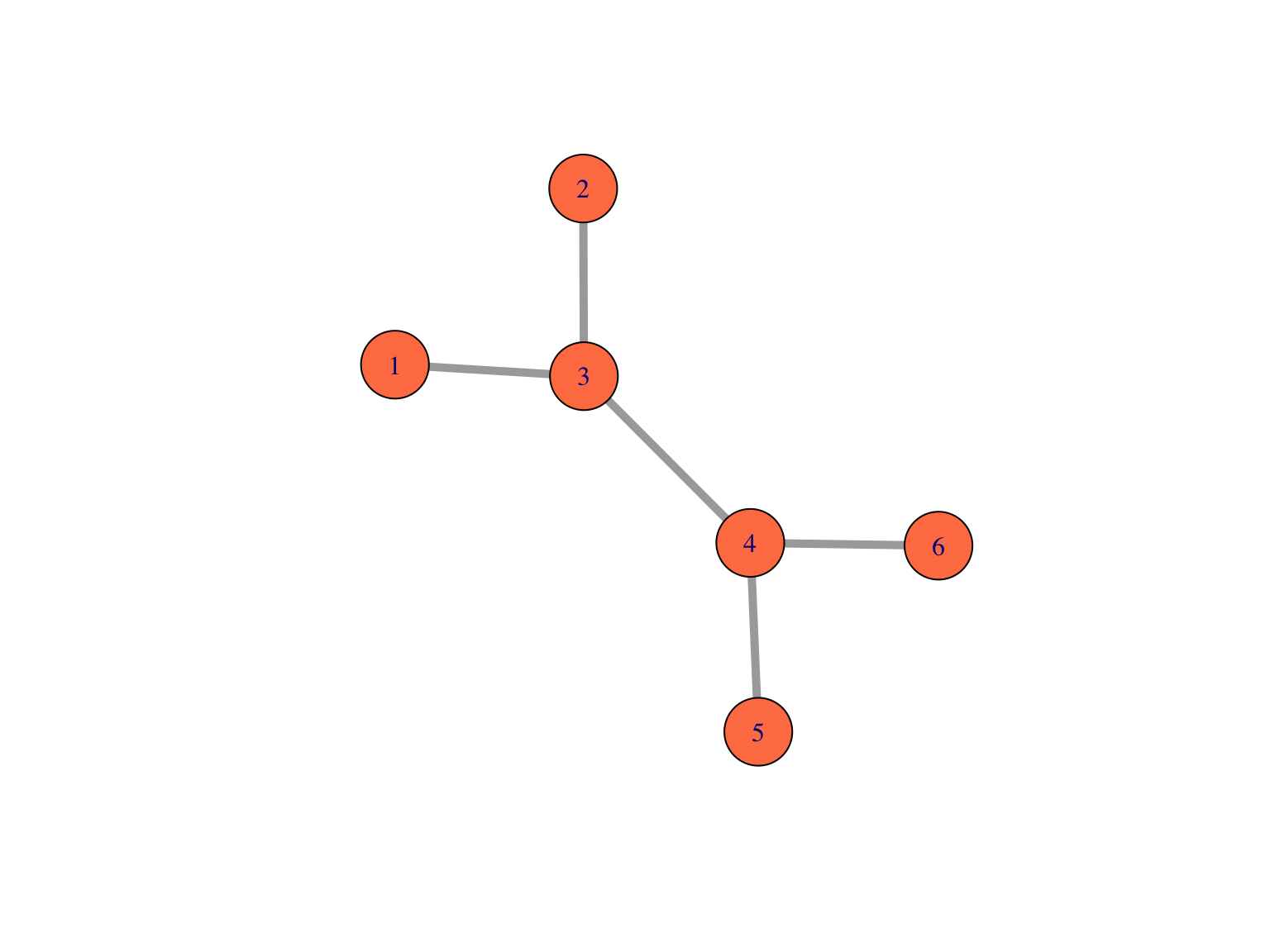
A small undirected graph with numbered nodes.
Here is the adjacency matrix of the small undirected graph represented below. We see that \(A\) is symmetric \(n \times n\) matrix of \(0\)’s and \(1\)’s.
A=as_adj(g1, sparse = FALSE)
A## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 0 0 1 0 0 0
## [2,] 0 0 1 0 0 0
## [3,] 1 1 0 1 0 0
## [4,] 0 0 1 0 1 1
## [5,] 0 0 0 1 0 0
## [6,] 0 0 0 1 0 0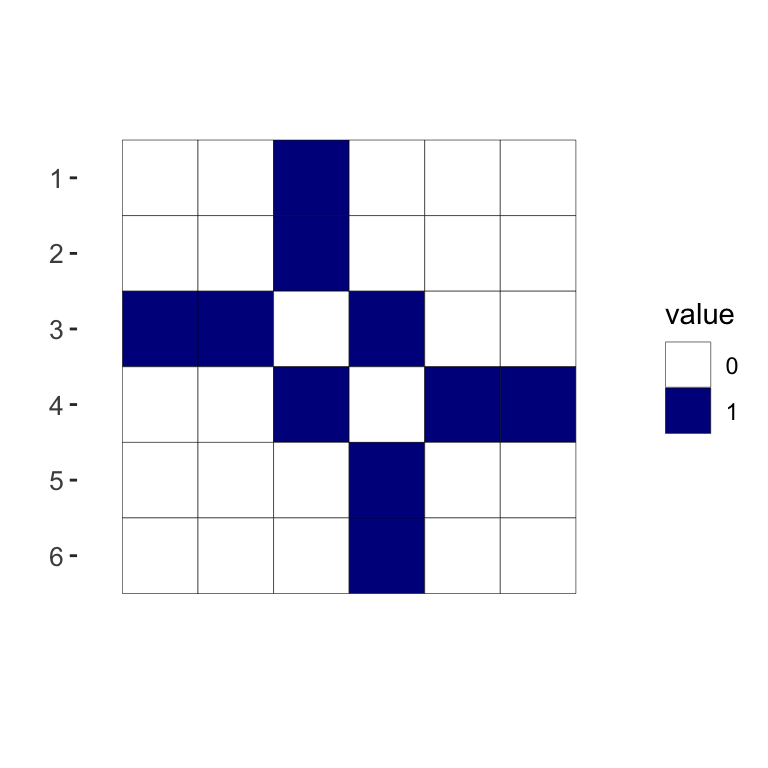
Here is how we plot a simple graph:
library("igraph")
edges1 = matrix(c(1,3,2,3,3,4,4,5,4,6), byrow = TRUE, ncol = 2)
g1 = graph_from_edgelist(edges1, directed = FALSE)
plot(g1, vertex.size = 25, edge.width = 5, vertex.color = "coral")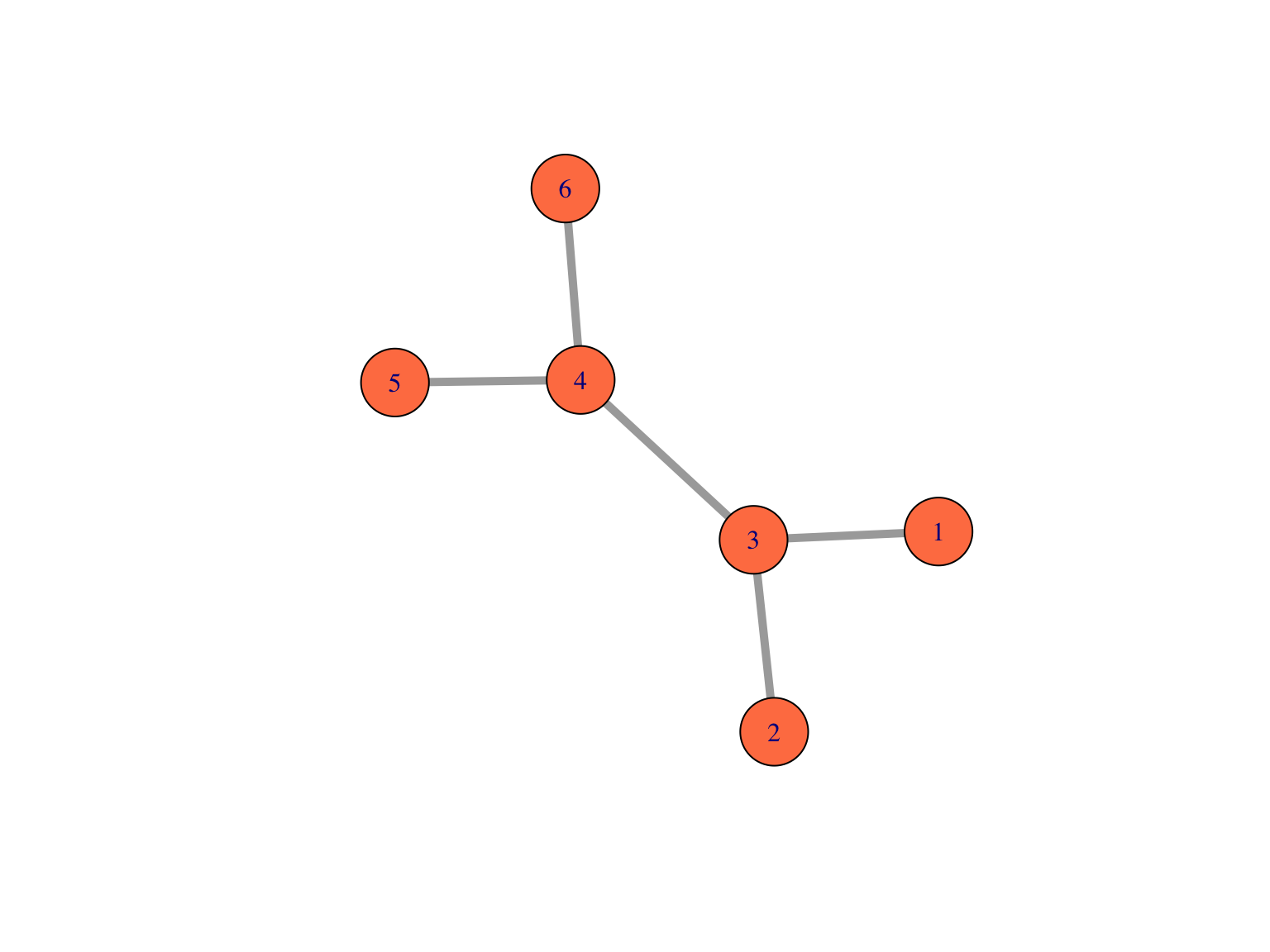
Now add two lines that label the nodes:
library("igraph")
edges1 = matrix(c(1,3,2,3,3,4,4,5,4,6), byrow = TRUE, ncol = 2)plot(sg, vertex.size = 35, edge.width = 7, vertex.color = "purple",vertex.label.color="white")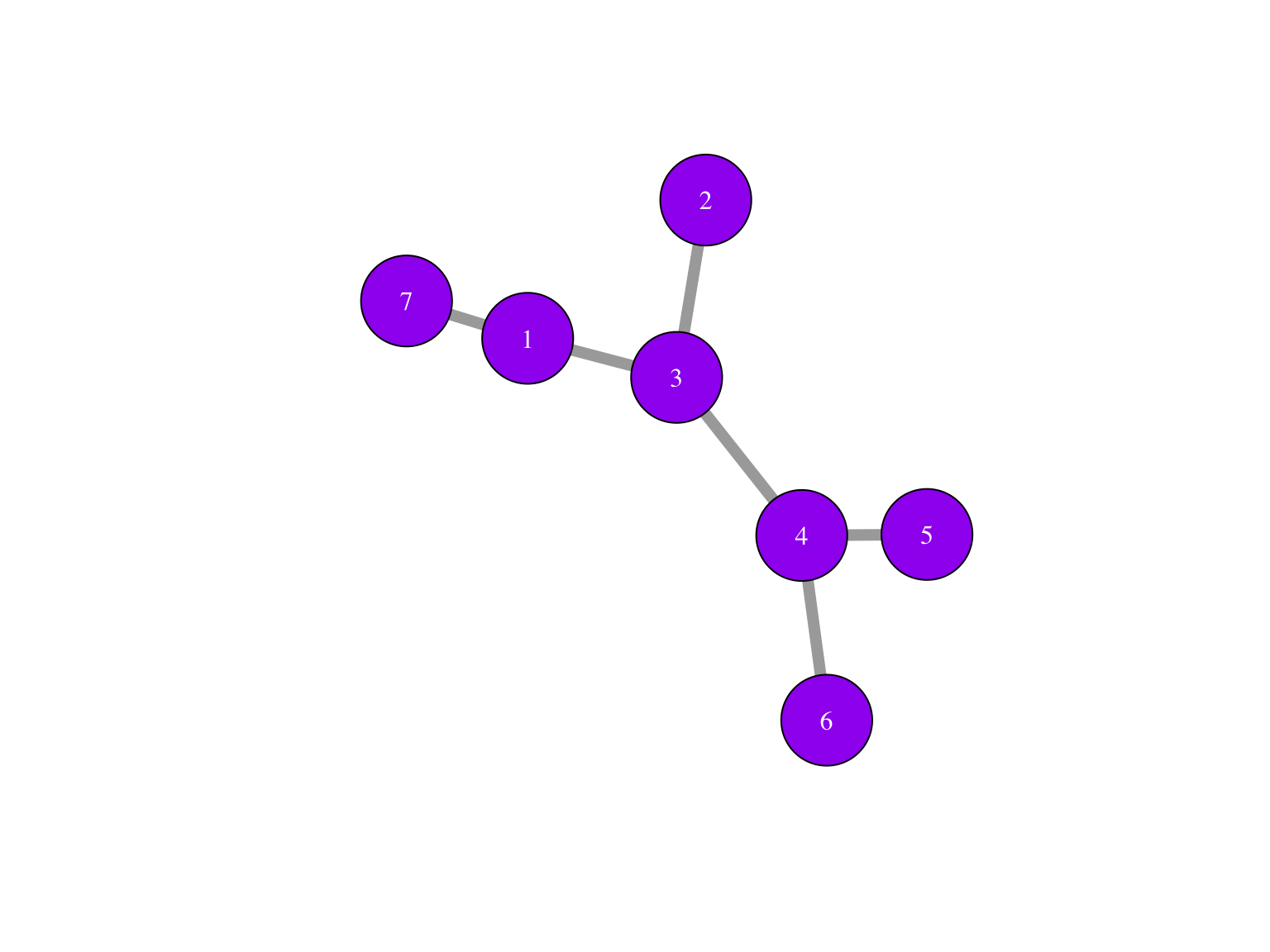
The nodes or vertices which are the colored circles with numbers in them in the Figure.
Edges or connections, the segments that join the nodes and which can be directed or not.
Edge lengths, when not specified, we suppose they are all one and compute the distance between vertices on the graph as the number the edges traversed. On the other hand, in many situations we have meaningful edge lengths or strengths of connections between vertices that we can use both in the plots and analyses.
The most common form of network that we create from data comes from some measure that we have available between the points that will be vertices of the graph. Suppose we are provided with a dist object from a distance computation between samples or nodes. There are several ways a graph can be constructed from this starting point.
library("igraph")
library("ggnetwork")
library("ggplot2")
pts = structure(c(0, 0, 1, 1, 1.5, 2, 0, 1, 1, 0, 0.5, 0.5),
.Dim = c(6L,2L))
matxy = pts
distxy = stats::dist(matxy)
g = graph.adjacency(as.matrix(distxy), weighted = TRUE)
mst1 = igraph::mst(g)A spanning tree is a tree that goes through all points at least once, the graph with red edges is such a tree.
The minimum spanning tree is the spanning tree with the shortest length.
Minimum spanning trees (MST) are easy/cheap to compute and there are many different functions in R packages that provide them:
igraph::mst ape::mst ade4::mstree vegan::spantree spdep::mstree
An example distance matrix was created between strains of HIV from different patients whose countries were recorded.
We can read in the DNA distance data that was provided.
Using ggnetwork and ape::mst
library(ggplot2)
library(igraph)
library(ggnetwork)
load(url("https://web.stanford.edu/class/bios221/data/dist2009c.RData"))
country09 = attr(dist2009c, "Label")
mstree2009 = ape::mst(dist2009c)
gr09 = graph.adjacency(mstree2009, mode = "undirected")
gg = ggnetwork(gr09, arrow.gap = 0, layout = layout_with_fr(gr09))
ggplot(gg, aes(x = x, y = y, xend = xend, yend = yend)) +
geom_edges(color = "black", alpha = 0.5, curvature = 0.1) +
geom_nodes(aes(color = name), size = 2) + theme_blank() +
geom_nodetext(aes(label = name), color = "black", size = 2.5) +
theme(plot.margin = unit(c(0, 1, 1, 6), "cm"))+
theme(legend.position = c(0, 0.14),
legend.background = element_blank(),
legend.text = element_text(size = 7))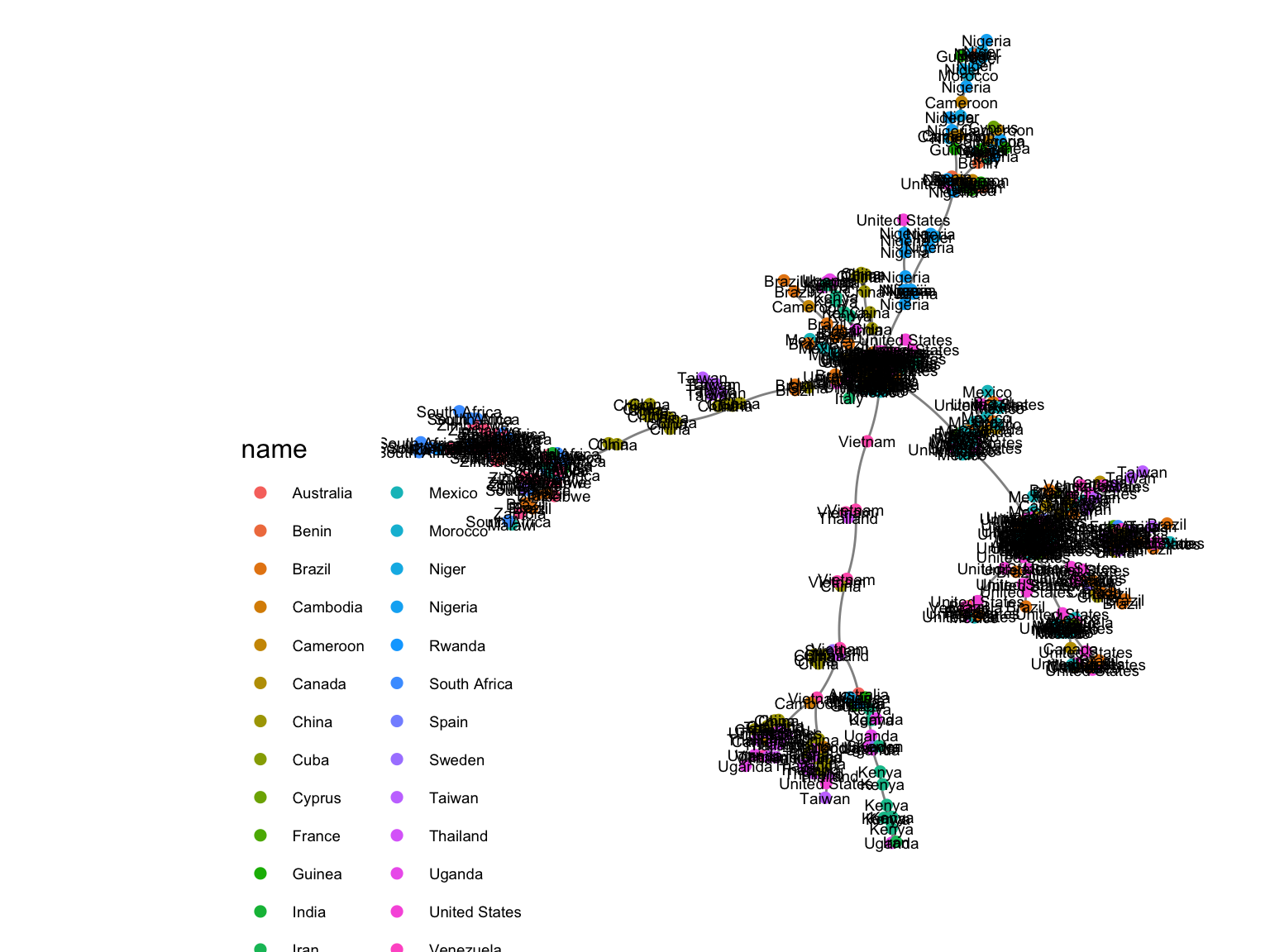
We can use the ggrepel package to make it cleaner:
ggplot(gg, aes(x = x, y = y, xend = xend, yend = yend)) +
geom_edges(color = "black", alpha = 0.5, curvature = 0.1) +
geom_nodes(aes(color = name), size = 2) +
geom_nodetext_repel(aes(label = name), color="black", size = 2) +
theme_blank() +
guides(color = guide_legend(keyheight = 0.3, keywidth = 0.3,
override.aes = list(size = 6), title = "Countries"))
tidygraph and ggraphMore recently, there has been a new development with an extension of the ggraph package to include tidyverse compatible data structures, through the tidygraph package which creates “graph” tibbles.
library(tidygraph)
library(ggraph)
# Make sure you have the data loaded
# load(url("https://web.stanford.edu/class/bios221/data/dist2009c.RData"))
country09 <- attr(dist2009c, "Label")
class(dist2009c)## [1] "dist"graph09 <- graph_from_adjacency_matrix(as.matrix(dist2009c), weighted=TRUE)
mstree2009tidy <- igraph::mst(graph09)%>%as_tbl_graph()
## Without the labels
ggt<-ggraph(mstree2009tidy, layout = 'nicely') + geom_edge_link() +
theme_graph()
ggt
## Adding labels
ggt+
geom_node_text(aes(label = name), colour = 'purple', vjust = 0.4)+ theme_graph()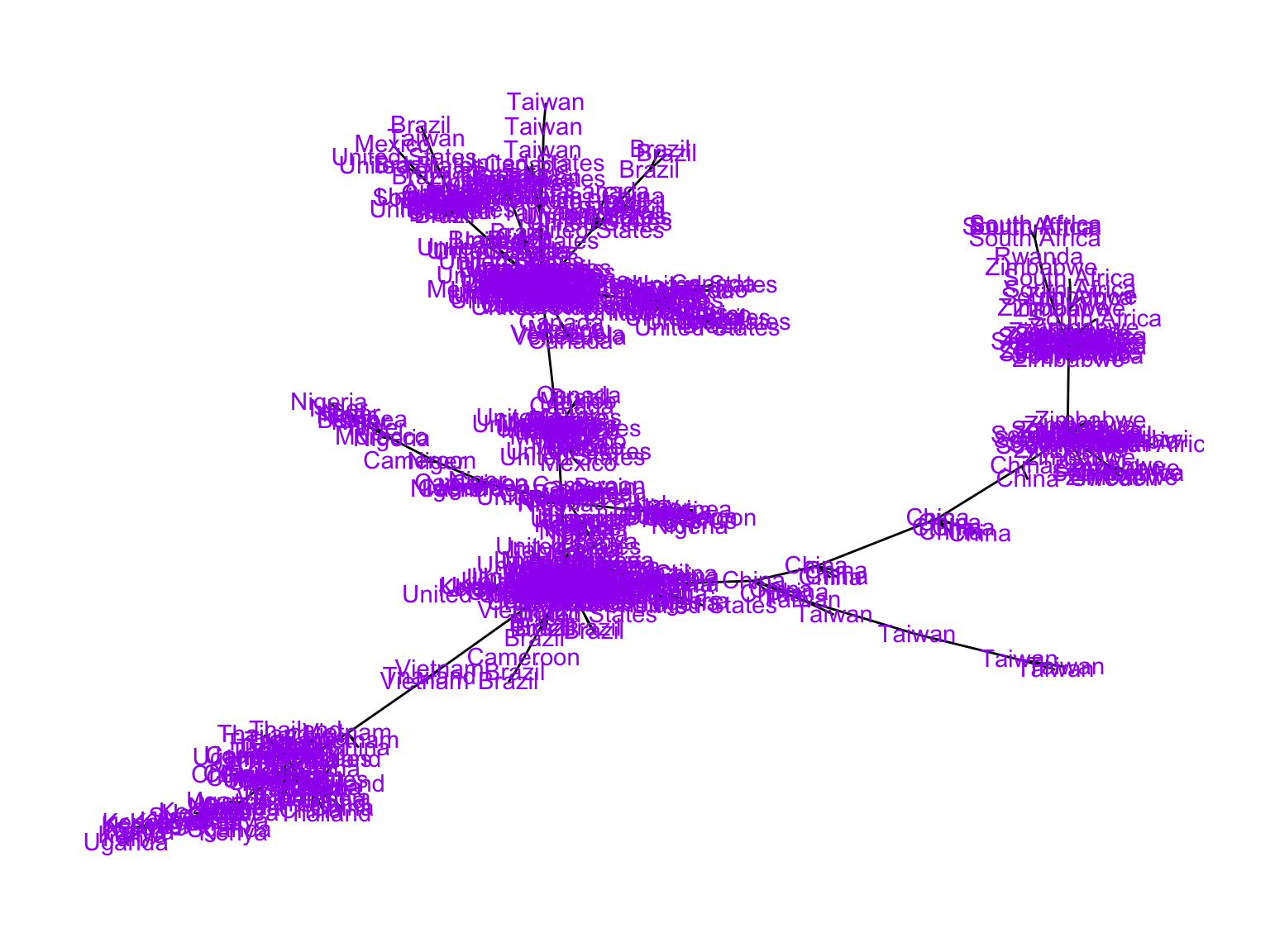
## Adding colors for countries
ggt+
geom_node_point(aes(color = name), size = 2) +theme_graph()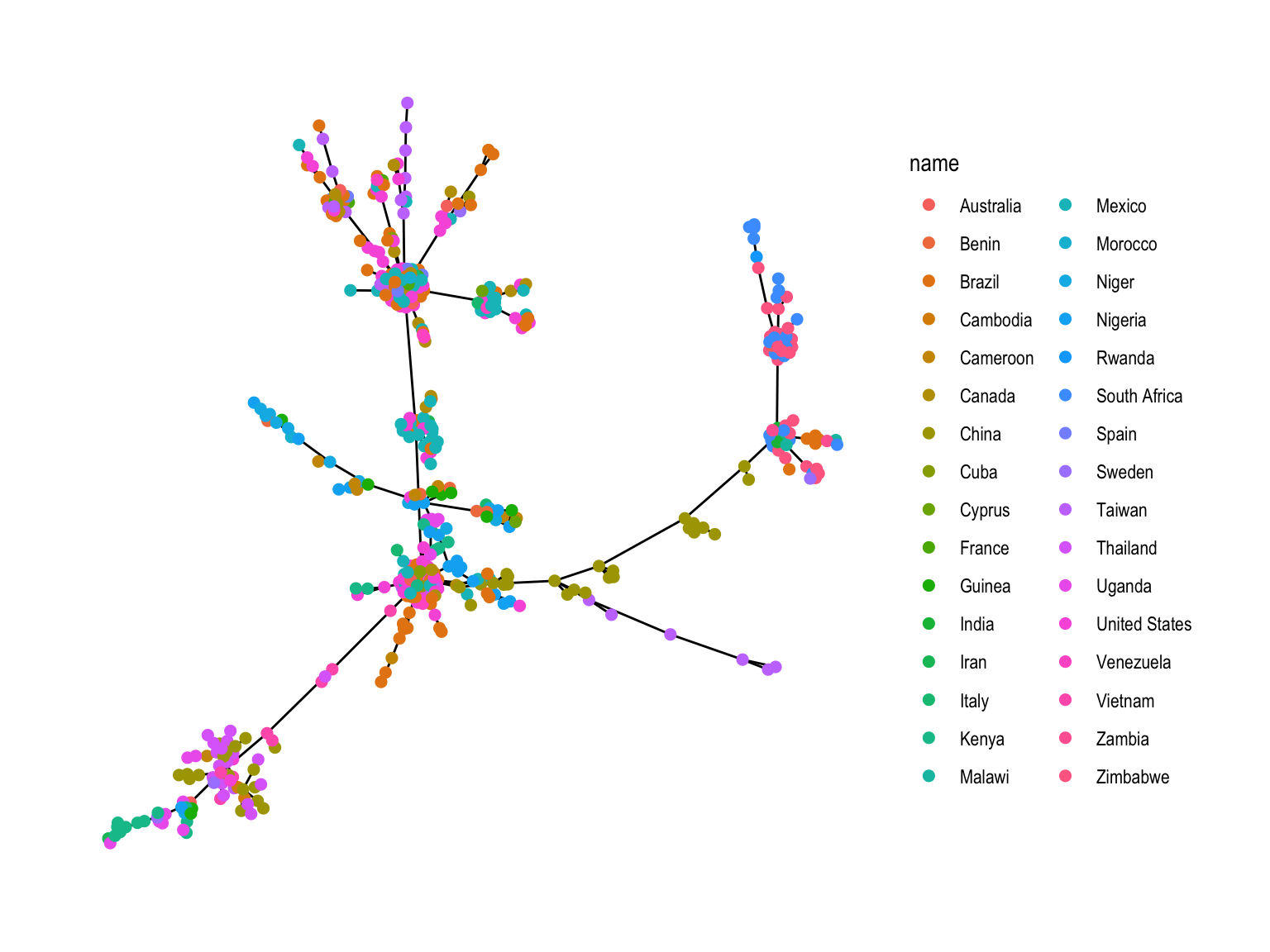
We would like to label only the countries which appear as hubs (ie with a high degree, say larger than 5).
We are going to compute the degrees and label the nodes according to their degrees. Let’s look at how to add that variable:
mstDegreed<-mstree2009tidy %>%
activate(nodes) %>%
mutate(degree=centrality_degree(,mode="all"))
mstDegreed## # A tbl_graph: 512 nodes and 511 edges
## #
## # A rooted tree
## #
## # Node Data: 512 x 2 (active)
## name degree
## <chr> <dbl>
## 1 Thailand 1
## 2 Mexico 1
## 3 Zimbabwe 1
## 4 Taiwan 1
## 5 India 1
## 6 Mexico 1
## # … with 506 more rows
## #
## # Edge Data: 511 x 3
## from to weight
## <int> <int> <dbl>
## 1 7 145 0.0375
## 2 7 213 0.0269
## 3 7 228 0.0402
## # … with 508 more rowsTry to modify the plotting code so that the labels of the higher degree hubs are larger and the graph nodes are readable:
ggraph(mstDegreed,layout = 'nicely') + geom_edge_link() +
geom_node_point(aes(color = name))+
geom_node_text(aes(label = name, size=degree))+
theme_graph()ggraph(mstDegreed,layout = 'nicely') + geom_edge_link() +
geom_node_point(aes(color = name))+
geom_node_text(aes(label = name, size=degree,alpha=degree),nudge_x=-13,nudge_y=-6.5)+
theme_graph()We are going to make a minimum spanning tree between HIV cases using the geographic location of each case was made to reduce overlapping of points; the DNA distances between patient strains were used as the input to an undirected minimum spanning tree algorithm, the world coordinates come from the rworldmap package.
library("rworldmap")
mat <- match(country09, countriesLow$NAME)
coords2009 = data.frame(
lat = countriesLow$LAT[mat],
lon = countriesLow$LON[mat],
country = country09)
layoutCoordinates = cbind(
x = coords2009$lon,
y = coords2009$lat)
ggt<-ggraph(mstree2009tidy, layout = layoutCoordinates) +
geom_edge_link() +
theme_graph()
ggt
ggt<-ggraph(mstree2009tidy, layout = layoutCoordinates) +
geom_edge_link(alpha=0.3) +
theme_graph()
ggt
When comparing these two graphs, what do we notice?
Alot of overlapping edges that are invisible.
How can we fix this?
One way is to use jitter for each of the points.
Try modifying the code to add a jitter and plot a more instructive version of the graph.
jitterlayoutCoordinates = cbind(
x = jitter(coords2009$lon, amount = 10),
y = jitter(coords2009$lat, amount = 7))
ggt<-ggraph(mstree2009tidy, layout = jitterlayoutCoordinates) +
geom_edge_link(alpha=0.5,linemitre=5) +
theme_graph()We actually need to keep the jittered coordinates and assign the labels to the nodes so they appear on the plot.
labc = names(table(country09)[which(table(country09) > 1)])
matc = match(labc, countriesLow$NAME)
dfc = data.frame(
latc = countriesLow$LAT[matc],
lonc = countriesLow$LON[matc],
labc)
ggt <- ggraph(mstree2009tidy, layout = jitterlayoutCoordinates) +
geom_edge_link(alpha=0.5,linemitre=5) +
geom_label(data = dfc, aes(x = lonc, y = latc, label = labc, fill = labc), colour = "white", alpha = 0.7, size = 3) +
geom_node_point(aes(color = country09), size = 2,alpha=0.4) + guides(fill=FALSE)+
theme_graph()
ggt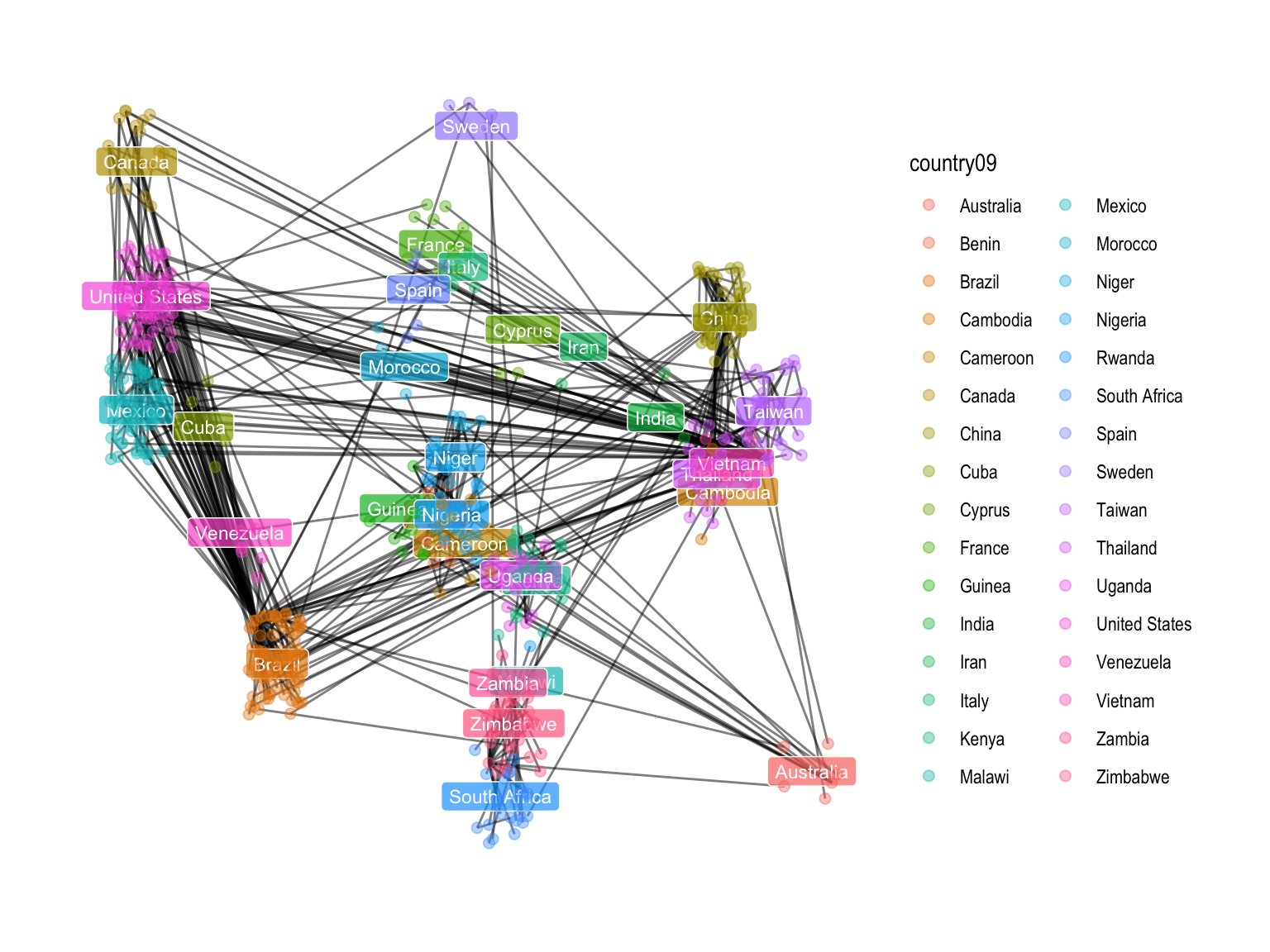
Try changing the type of edge (bend,diagonal,arc)
ggt <- ggraph(mstree2009tidy, layout = jitterlayoutCoordinates) +
geom_edge_link(alpha=0.5,linemitre=5) +
geom_label(data = dfc, aes(x = lonc, y = latc,label = labc, fill = labc), colour = "white", alpha = 0.7, size = 3) +
geom_node_point(aes(color = country09), size = 2,alpha=0.4) +
guides(fill=FALSE)+
theme_graph()
ggtggt <- ggraph(mstree2009tidy, layout = jitterlayoutCoordinates) +
geom_edge_bend(alpha=0.5,linemitre=5) +
geom_label(data = dfc, aes(x = lonc, y = latc,label = labc, fill = labc), colour = "white", alpha = 0.7, size = 3) +
geom_node_point(aes(color = country09), size = 2,alpha=0.4) +
guides(fill = FALSE) +
theme_graph()
ggtAnother new possibility is the new sfnetworks package which allows for paths and routing
For univariate data in two groups, we can test the differences by looking at the number of runs within each group.
Seeing the number of runs in a one-dimensional, two-sample, nonparametric Wald-Wolfowitz test can indicate whether the two groups have the same distributions.
The nodes in the graph correspond to the samples and are associated to factors that specify the litter and individual ids of the mice from which the samples were collected.
library("phyloseq")
library("phyloseqGraphTest")
library("igraph")
## data(ps1)
ps1 = readRDS(url("https://web.stanford.edu/class/bios221/data/ps1.rds"))
ps1## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 389 taxa and 344 samples ]
## sample_data() Sample Data: [ 344 samples by 16 sample variables ]
## tax_table() Taxonomy Table: [ 389 taxa by 6 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 389 tips and 387 internal nodes ]sampledata = data.frame( sample_data(ps1))
d1 = as.matrix(phyloseq::distance(ps1, method="jaccard"))
gr = graph.adjacency(d1, mode = "undirected", weighted = TRUE)
net = igraph::mst(gr)
V(net)$id = sampledata[names(V(net)), "host_subject_id"]
V(net)$litter = sampledata[names(V(net)), "family_relationship"]The minimum spanning tree based on Jaccard dissimilarity and annotated with the mice ID and litter factors.
library(ggnetwork)
gnet=ggnetwork(net)
ggplot(gnet, aes(x = x, y = y, xend = xend, yend = yend))+
geom_edges(color = "darkgray") +
geom_nodes(aes(color = id, shape = litter)) + theme_blank()+
theme(legend.position="bottom")
library("phyloseqGraphTest")
gt = graph_perm_test(ps1, "host_subject_id", distance="jaccard",
type="mst", nperm=1000)
plot_test_network(gt)
gt## Output from graph_perm_test
## ---------------------------
## Observed test statistic: 149 pure edges
## 343 total edges in the graph
## Permutation p-value: 0.000999000999000999plot_permutations(gt)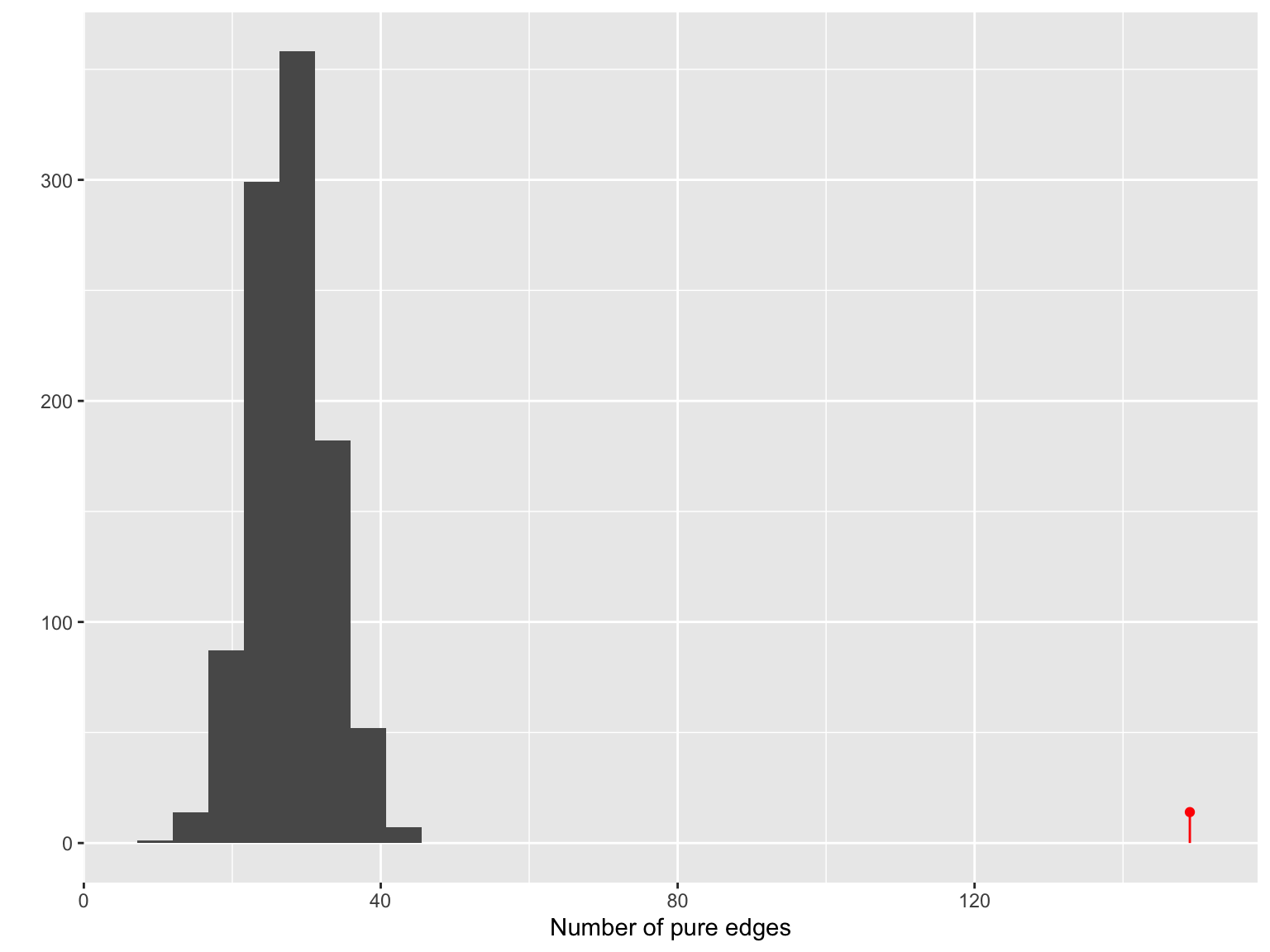
gt$pval## [1] 0.000999001The permutation histogram of the number of pure edges in the network obtained from the minimal spanning tree with Jaccard similarity.
net = make_network(ps1, max.dist = 0.35)
sampledata = data.frame(sample_data(ps1))
V(net)$id = sampledata[names(V(net)), "host_subject_id"]
V(net)$litter = sampledata[names(V(net)), "family_relationship"]
netg = ggnetwork(net)ggplot(netg, aes(x = x, y = y, xend = xend, yend = yend)) +
geom_edges(color = "darkgray") +
geom_nodes(aes(color = id, shape = litter)) + theme_blank()+
theme(plot.margin = unit(c(0, 5, 2, 0), "cm"))+
theme(legend.position = c(1.4, 0.3),legend.background = element_blank(),
legend.margin=margin(0, 3, 0, 0, "cm"))+
guides(color=guide_legend(ncol=2))
A co-occurrence network created by using a threshold on the Jaccard dissimilarity matrix. The colors represent which mouse the sample came from; the shape represents which litter the mouse was in.
gt = graph_perm_test(ps1, "family_relationship",
grouping = "host_subject_id",
distance = "jaccard", type = "mst", nperm= 1000)
gt$pval## [1] 0.000999001plot_permutations(gt)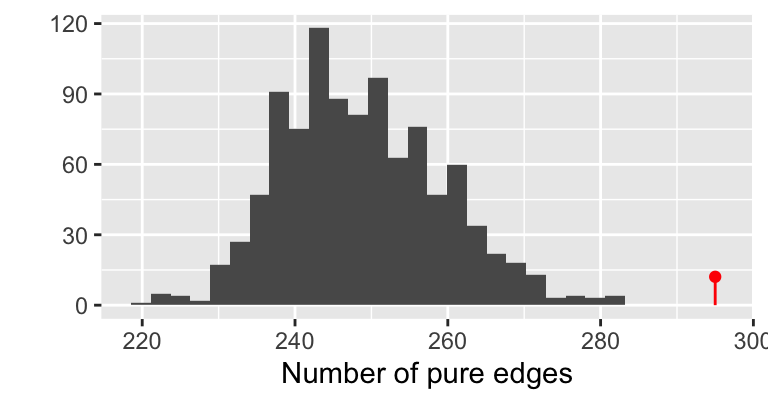
The permutation histogram obtained from the minimal spanning tree with Jaccard similarity.
gtnn1 = graph_perm_test(ps1, "family_relationship",
grouping = "host_subject_id",
distance = "jaccard", type = "knn", knn = 1)
gtnn1$pval## [1] 0.004plot_test_network(gtnn1)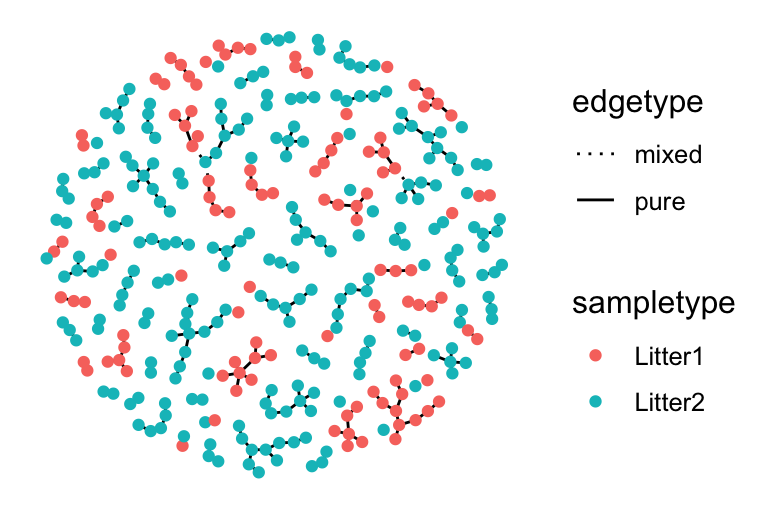
The graph obtained from a nearest-neighbor graph with Jaccard similarity.
phyloseq network
tidygraph_hex.png