
Genetic Modifiers of Huntington’s Disease
Explaining the variable onset, manifestation, and progression of HD
Huntington’s disease (HD) is caused by an expansion in the Huntington gene, which codes for the huntingtin protein. This gene contains a repeated span of three nucleotides, C-A-G, that encode for the amino acid glutamine. Individuals with an increased number of CAG repeats in the HD gene thus produce a mutated version of the huntingtin protein which contains too many glutamines. Although the exact function of the huntingtin protein is not known, the mutated protein is thought to be responsible for the widespread neurodegeneration that takes place in HD.
Since the HD gene was first characterized in 1993, scientists have known that the number of CAG repeats is related to whether an individual will develop the disease. In fact, the genetic test for HD relies on measuring the number of CAG repeats and determining based on this repeat length if an individual is gene positive, gene negative, or in the intermediate range where symptoms may not occur in an individual’s lifetime. Additionally, CAG repeats have also been shown to be inversely correlated with the age of onset in HD. This means on average, a person with more CAG repeats in the HD gene will have an earlier age of onset than a person with fewer CAG repeats in the HD gene.
However, there are several important caveats to be mentioned about the relationship between CAG repeats and HD. For one, the correlation between CAG repeat number and age of onset is significantly weaker for individuals with a shorter number of repeats. Among individuals with between 40 and 50 repeats (an estimated 90% of people affected by HD worldwide), CAG repeat length has been statistically shown to only account for approximately 44% of the variation seen in the age of onset. For these individuals, other factors contribute to the age of onset for HD. Thus, although a person with a longer CAG repeat number is on average more likely to get HD earlier in life, this is by no means a certainty, especially for individuals with a lower number of repeats. For any given CAG repeat length between 40 and 50, there is a large range in the age of onset for HD—indeed, two people from the same family with the same CAG repeat number might present symptoms of HD more than 20 years apart. This observation suggests that other factors besides CAG repeat number likely affect the age of onset as well.
The action of genetic modifiers can help explain some of the variable onset, manifestation, and progression of HD. Genetic modifiers are any genes that affect the phenotype, or physical manifestation, of another gene. In the case of genetic diseases such as HD, you can think of genetic modifiers as different seasonings that change the flavor of a particular dish (i.e. the huntingtin gene)—regardless of the original composition of the dish, the final flavor can be better or worse depending on what seasoning you add and how much of it you use. Likewise, the progression of HD is likely to be at least partially dependent on the interactions between the mutated HD gene (the “original dish”) and the particular combination of genetic modifiers (the “seasonings”). The variable actions of genetic modifiers can help explain why two people with the exact same number of CAG repeats can have vastly different onset times, manifestations and progressions of the disease.
Identifying modifier genes^
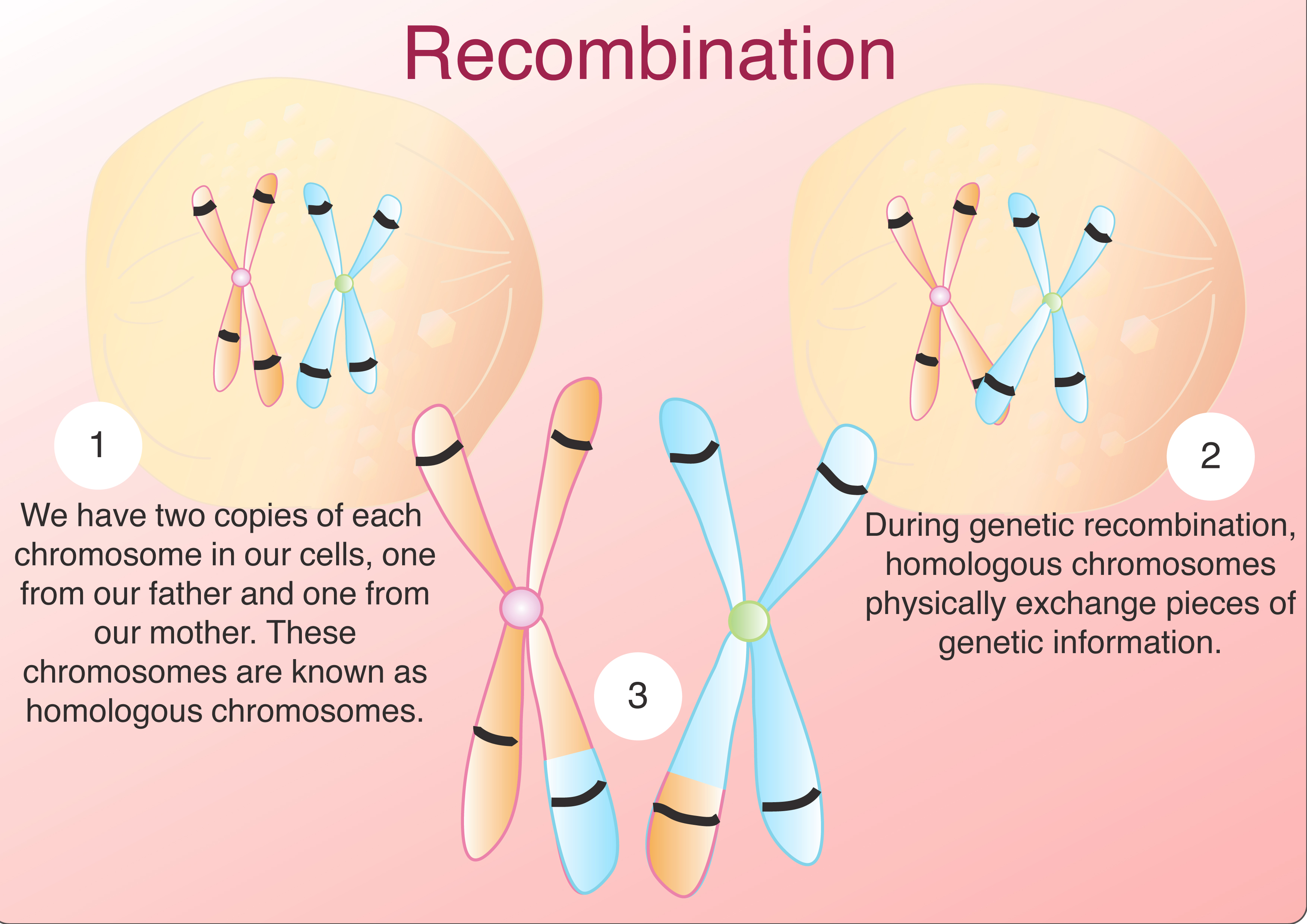
Although there are several different strategies available to find genetic modifiers, almost all of them are related to the idea of genetic linkage. Genetic linkage is based on the principle that human chromosomes exchange pieces of genetic material during meiosis in a process known as recombination (Click here for more information on meiosis and recombination). Homologous chromosomes are pairs of chromosomes, one originating from each parent, with corresponding DNA sequences. Humans have 23 chromosome pairs. During genetic recombination, homologous chromosomes physically exchange pieces of genetic information. Since one of these homologous chromosomes came from the mother and the other came from the father, recombination is one way for organisms to promote genetic diversity. When the sister chromatids later separate during the second stage of meiosis, they become the genetic material for two different gametes (sperm or egg cells). But these sequences of genetic information are not simple copies of maternal and paternal DNA. Instead, thanks to the recombination that took place in meiosis, the DNA of these gametes contain new arrangements of genetic information. Because recombination involves the physical exchange of genetic information, genes that are closer together are more likely to stay together through meiosis than genes that are further apart as it is less likely the homologous chromosomes will exchange material in the short distance between them.
If there is enough genetic data from multiple generations of a family, linkage analysis can provide a powerful tool for the identification of genetic modifiers of HD. To look for genetic modifiers, scientists look for specific genes and even gene arrangements that are associated with, or linked, to a specific aspect of the disease. For example, if a marker on gene A has been shown to be frequently present in individuals with a particularly severe manifestation of cystic fibrosis, we can suspect this marker is somehow linked to the gene or genes responsible for this manifestation of cystic fibrosis. We can then use statistical tools to determine if gene A is indeed linked to severe cases of cystic fibrosis.
Modifier genes and HD^
Scientists have looked for genes that are linked to the age of onset for HD. Recall that one major predictor of the age of onset is the number of CAG repeats (see introduction for more information). After researchers accounted for the differences due to CAG repeat length, they found strong evidence that a region of DNA on chromosome 6 was linked to the age of onset of HD in individuals with the mutant HD gene, even though the gene for HD is found on chromosome 4. If the results of this linkage analysis are correct, they would suggest that a gene (or group of genes) within this region on chromosome 6 act as genetic modifiers affecting the progression of HD in appreciable ways. A similar study on the population of affected individuals in Lake Maracaibo, Venezuela, backed up these findings and identified two additional regions on the chromosome that were linked to HD. These spans of the genome that may be genetic modifiers are rather large—for example, one of the candidate regions in chromosome 6 encompasses about 128 genes that are either known to be there, or predicted to be present. As a result, it is difficult to identify the specific gene that is a relevant genetic modifier in HD. The next step would then be to narrow down this large list of candidates by looking at how the protein products of these genes are affected in individuals with HD. Eventually, the goal would be to identify specific genes that act as genetic modifiers for age of onset of the disease.
But even if a genetic modifier for HD is successfully isolated and identified, there will still be a significant amount of work to be done. For one, researchers must be able to explain how this gene affects the manifestation and progression of HD. What proteins are targeted? What pathways are involved? Although these questions will not be easy to answer, the ability to do so will not only further scientific understandings of how HD progresses, but can also be of great use in the development of future treatments or therapies. If we were able to identify a gene (or a combination of genes) that makes the onset, manifestation, or progression of HD worse, we could potentially develop drugs or therapies that target the harmful effects of this modifier gene. The identification and characterization of genetic modifiers of HD holds great promise for future research.
Sources^
Génin E., Feingold, J. & Clerget-Darpoux, F. (2008). Identifying modifier genes of monogenic disease: strategies and difficulties. Human Genetics 124: 357-368.
This article reviews different methods for identifying the genetic modifiers for single-gene diseases
Gusella, J.F. & MacDonald, M.E. (2009). Huntington’s disease: the case for genetic modifiers. Genome Medicine 1: 80:1-6.
This article, while technical, effectively summarizes the current research on genetic modifiers of HD
Li, J.L. et al. (2006). Genome-wide significance for a modifier of age at neurological onset in Huntington’s Disease at 6q23-24: the HD MAPS study. BMC Medical Genetics 7: 71.
This primary source article presents the results of a large-scale genome scan that found evidence for a genetic modifier of HD on chromosome 6
Gayán, J. et al. (2008). Genomewide linkage scan reveals novel loci modifying age of onset of Huntington’s disease in the Venezuelan kindreds. Genetic Epidemiology 32: 445-453.
This study used genomic scanning of individuals from Lake Maracaibo to identify several candidates for genetic modifiers of HD.
Y. Lu 2011