
PRIONS FACT SHEET
- Prions are not viruses and unlike any living pathogens because they don’t have a genome/nucleic acids. Since different strains or prions have the same or extremely similar amino acid sequences, strainsare distinguished by differences in conformation and glycosylation.
- Infectious particle PrP(sc) has the same amino acid sequence as PrP(c), which is a naturally occurring, constitutively expressed protein found mostly in the host’s neurons. PrPsc and PrPc differ in secondary structure, folding conformation, protease resistance, resistance to denaturation, and aggregability.
- “Replication” of PrPsc occurs when PrPsc induces PrPc to change conformation to another isoform, PrPsc. PrPsc consist of 40% alpha-helix structures and very little beta-sheet structures, and PrPc has 30% alpha-helix structure and 45% beta-sheets.
- PrPsc is necessary for prion infection; without PrPsc, infectious PrPc cannot be made. A species barrier exists because PrPc infection is easier when the PrPc and target PrPsc sequences are the same.
- Once an individual is infected and neurologic signs appear, no documented patient has recovered. The host cannot mount an immune response against it because PrPSc and PrPc have the same epitopes.
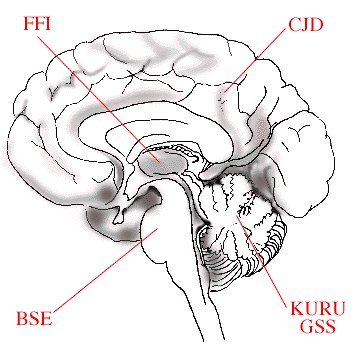
- Prions accumulate in the central nervous system, when host clearance is slower than the rate of PrPsc formation. Thefigure on the right shows how different human prion diseases affect different brain regions. Fatal familial insomnia(FFI) mainly affects the thalamus region, Bovine Spongiform Encephalophaty (BSE) - the brain stem, Kuru and Gerstmann-Straussler-Scheinker (GSS) - the cerebellum, and Creutzfeldt-Jakob Disease (CJD) - the cerebrum. (http://nobelprize.org/nobel_prizes/medicine/laureates/1997/press.html)
- Although it receives the most media coverage, “mad cow disease,” or variant Creutzfeld-Jakob disease, accounts for less than 1% of all human prion infections. (That < 1% also includes iatrogenic CJD from hGh, corneal transplants, blood transfusions, and dura mater grafts). 85-90% occur spontaneously, and a genetic link is suspected for the remaining 10-15%.
- Very long incubation period, on the scale of years (4-30 years for vCJD), but relatively short duration of illness (average of 13 months, vCJD).
- Cytopathic effect of PrP amyloid plaques with severe spongiform degeneration of the gray matter in the brain that gives it a “Swiss cheese” or “spongy” look (hence the name bovine “spongiform” encephalopathy). Other pathology includes prominent neuronal loss, atypical brain inflammation, synaptic dysfunction, and other forms of prion protein aggregates in the cerebrum.
- Mechanisms to convert PrPsc into PrPc and for prion infection to enter the nervous system have not been confirmed. Several hypotheses for molecular mechanism of conversion include the template-assisted model, the nucleation/polymerization model, and the assisted-nucleation model. PrPc could infect the CNS after entry via peripheral nerves, the bloodstream, or a combination.
Return to Main Menu