
SMALL BUT DEADLY
Prion researcher Stanley Prusiner aptly coined the name “prions,” short for “proteinaceous infectious particles.” Prions are infectious proteins found in mammals and fungi that “infect” host organisms by inducing a host precursor protein PrP(c) to change conformation into a disease causing isoform (PrP(sc)). Prions are different from viruses because they have no nucleic acid genome, are constructed of only protein, do not require exogenous infection (most infections in humans are spontaneous) and are nonimmunogenic. Nonimmunogenicity arises because PrP(c) and PrP(sc) share the same epitopes, and the host’s immune system recognizes PrP(c) as "self". However, similar to viruses, prions have a narrow host range, can cause exogenous infection, have cellular tropism, cause cytopathic effects, and generally have a clinical trajectory.
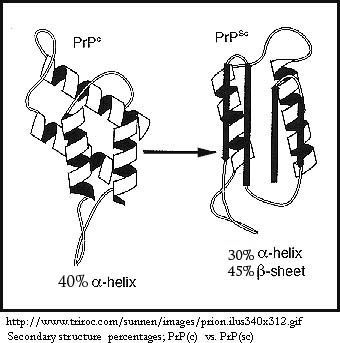
There are different strains of PrP “prion protein” that vary between species. Unlike viruses, strains can't be distinguished by differences in nucleic acid sequence, so differences in glycosylation and conformation are used as defining characteristics. Differences in strain explain why a species barrier exists, and cross-species infection is much more common between animals that are more evolutionarily related. PrP(c) is constitutively expressed in many tissues in the brain region and is expressed at the highest concentration in neurons. PrP(c) and the corresponding PrP(sc) that “infects” that specific strain of PrP(c) have identical or very near identical amino acid sequences, differing by a single- to-a few amino acids. Secondary protein structure differs dramatically, however, and the change in conformation also confers different properties, as shown in the chart below:
|
PrP(c) |
PrP(sc) |
Secondary structure |
More alpha-helix structure and less beta-sheet content (40%; little) |
Lless α-helix structure and more β-sheet content (30%; 45%) |
Cellular expression/ tropism |
Constitutively expressed in central nervous system, lymphoid, follicular dendritic cells; lower levels in stomach, spleen, and kidneys |
Only neurons, myocytes, follicular dendritic cells, and B-lyphnocytes support formation |
Protease resistance |
Lower |
Higher |
Solubility |
Higher |
Lower |
Resistance to denaturation |
Lower |
Higher |
Aggregability |
None |
High levels |
Temperature labile |
More |
Less |
Normal PrP(c) Function
Despite its importance in prion infection, the function of PrP(c) is largely unknown. Given its high level of conservation across mammals, PrP(c) is suspected to have an important biological function. Prp(c) is a glycosylphosphatidylinositol (GPI)-anchored cell-surface glycoprotein, and it is found in the lipid rafts of brain cells and neurons, a membrane region specialized in signaling. Its exact function is unknown, but PrP(c) may act in a neuroprotective role in signal transduction. PrP(c) may be involved in the binding and normal brain metabolism of copper, helping maintain copper homeostasis. Several observations support the copper-metabolism hypothesis: 1) if copper homeostasis isn’t maintained, neuronal damage results, 2) in scrapie-infected murine models, a significant decrease in brain copper levels were observed before onset of clinical symptoms (the same was observed in humans affected by sCJD), and 3) Copper binding makes beta-sheets more thermodynamically stable, so that the change from alpha-helices in PrP(c) to beta-sheets in PrP(sc) is favored. Because PrP(c) also interacts with with Bcl-2, a protein in the antiapoptosis family, it’s possible that PrP(c) is an antiapoptotic protein. But since it hasn’t been found in the cytoplasm, mitochondria, or endoplasmic reticulum where these proteins act, this is a less-likely hypothesis.
Formation of PrP(sc) is directly proportional to the level of PrP(c) expression in the brain and PrP(sc) that is in the body. PrP(c) is necessary for infection, however, because PrP(sc) needs host protein to replicate.
Brief overview of the pathogenesis of PrP(sc):
How PrP(sc) converts PrP(c) into an infectious form and what path ingested PrP(sc) takes to reach the brain are unknown, but several hypotheses are discussed here.
Mechanisms of PrP(c) --> PrP(sc) conversion:
I. Template-assisted conversion model
II. Nucleation/polymerization model
III. Assisted-nucleation model
Ingested Prions: Stomach to the Brain
PrP(sc) can enter the body through feeding, intravenous injection, intraperitoneally, or through the eye via corneal grafts and ocular injection. Though 85-90% of all human prion cases arise spontaneously and another 10-15% are familial, consumers are primarily concerned with “mad cow disease” or one form of variant Creutzfeldt-Jakob Disease caused by bovine prions. Human bovine spongiform encephalopathy (hBSE) is caused by ingesting contaminated beef products. How PrP(sc) can survive the acidity of the stomach and protease in the gut are unknown, but it does have a much higher resistance to both factors than PrP(c) does. It’s suspected that PrP(sc) enters the bloodstream throghthe distal ileum in the small intestine, through regions called Peyer’s patches. From there, PrP(sc) goes to the lymphoid tissues, where it’s hypothesized that follicular dendritic cells or B lymphocytes are targeted sites of replication.
From here, PrP(sc) could enter the brain from two distinct pathways: via peripheral nerves or via the bloodstream. Peripheral nerves are innervated by the sympathetic nervous system, which connects to the thoracic spinal cord of the central nervous system. Axonal and nonaxonal mechanisms are likely involved from here. PrP(sc) may be spread through direct exchange between plasma membranes of neighboring cells; this hypothesis would explain the long latency period of prion infection. However, since PrP(sc) infection is not localized to specific regions in the brain, the bloodstream hypothesis would help explain why multiple, unconnected regions are infected. Experiments have shown that it is likely a combination of both pathways.
Bibliography:
Bainbridge, John and Kenneth Barry Walker. (2005). The normal cellular form of prion protein modulates T cell responses. Immunology letters 96(1): 147-50.
CDC. (2007). Prevention of Specific Infectious Diseases. CDC Travelers’ Health: Yellow Book. http://wwwn.cdc.gov/travel/yellowBookCh4-VariantPrions.aspx
Collinge, John. (2001). Prion diseases of humans and animals: Their causes and molecular basis. Annual Review of Neuroscience 24: 519-50.
Fields Virology, 5th ed. (2007)
Medical Research Council Clinical Trials Unit. (2006). Prion-1: Randomised trial of quinacrine in human prion disease. http://www.ctu.mrc.ac.uk/studies/cjd.asp
Soto, Claudio. Prions: The New Biology of Proteins. Boca Raton, FL: Taylor & Francis, 2006.
Tuite, Mick F and Nadejda Koloteva-Levin. (2004). Propagating prions in fungi and mammals. Molecular Cell 14(5): 541-52.
Return to Main Menu