



|

|
We study the evolution of complex traits by developing new experimental and computational methods.
Although genetics is often taught in terms of simple Mendelian traits, most traits are far more complex. They evolve via a
multitude of genetic changes, each having a small effect by itself, which in sum give rise to the spectacular adaptation of every
organism to its environment.
Our work brings together quantitative genetics, genomics, epigenetics, and evolutionary biology to achieve a deeper understanding
of how genetic variation shapes the phenotypic diversity of life. Our main focus is on the evolution of gene expression, since this
is the primary fuel for natural selection. Our long-term goal is to understand the genetic basis of complex traits well enough to
introduce them into new species via genome editing.
Some specific projects in the lab
Exploring the genome with high-throughput precision genome editing
CRISPR has revolutionized our ability to edit genomes. Genome editing can either be precise— installing the exact desired edit—
or random. High-throughput CRISPR screens are possible, but have been restricted to introducing random mutations, severely
limiting their utility. To address this, we have developed CRISPEY, a CRISPR-based technology that enables genome editing that is
both high-throughput and precise. In our first project, we applied CRISPEY to measure the fitness effects of thousands of genetic
variants. This approach also allows us to explore interactions among genetic variants (epistasis) or with the environment (GxE),
and to map fitness landscapes with unprecedented resolution and throughput.
Publications: Sharon, Chen, Khosla et
al 2018.
Genome-wide scans for adaptive evolution
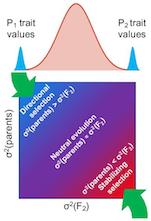
Natural selection is the force that drives evolutionary adaptation. However, not all traits are under selection; a key question
is which traits have been shaped by selection, as opposed to the random drift of neutral traits. We have developed methods to
distinguish between these, more easily and powerfully than was previously possible. This work has revealed that natural selection
on gene expression levels is widespread, often acting on the expression of entire pathways and protein complexes. We have found
these adaptations to affect traits including morphology, pathogenicity, and behavior. Most recently, we have applied these
methods to human/chimpanzee hybrid cells to reveal adaptive evolution of astrocyte-associated genes and hedgehog signaling. We
are currently extending these methods in a variety of ways, and applying them to a wide range of traits and species. Ultimately
we aim to understand the role of natural selection in the evolution of complex traits across the tree of life.
Publications: Fraser et al 2010; Fraser et al 2011;
Fraser 2011; Fraser et al 2012; Fraser 2013; Artieri & Fraser 2014; Naranjo et al 2015; Kita & Fraser 2016; Kita et al 2017; Artieri et al 2017; York et al 2018.
Uncovering the genetic basis of complex traits

The genetic basis of phenotypes is usually investigated with genetic mapping approaches known as QTL mapping or GWAS. These are
effective at finding genomic regions of interest, but they have several drawbacks: they do not implicate specific genes or
genetic variants; they do not reveal molecular mechanisms involved; and they require genotyping and phenotyping hundreds or
thousands of individuals. We are improving on these methods in two ways. First, we are developing new methods to map genetic
variants affecting molecular-level traits (such as DNA methylation and transcription factor binding) throughout the genome, which
do not require any individual-level genotyping or phenotyping. In addition, we are using inter-species hybrids to map the
landscape of cis-regulatory divergence, which allows us to identify the genes underlying polygenic adaptations far more
efficiently than QTL mapping. We are currently applying this approach to study adaptations in yeast, mice, primates,
sticklebacks, and more.
Publications: Fraser et al 2011;
Fraser 2011; Fraser et al 2012; Artieri & Fraser 2014; Kaplow et al 2015; Naranjo et al 2015; Tehranchi et al 2016; Tehranchi et al 2019.
|
|
