
Our group uses a wide range of spectroscopic methods coupled to calculations (density functional and ligand field theories) to understand the electronic structure of metal sites and its contribution to physical properties and reactivity. Much of our research effort focuses on understanding key problems in bioinorganic chemistry.
The bioinorganic systems studied in our group primarily fall into four areas: electron transfer sites, copper active sites in biology, mononuclear non-heme iron enzymes, and binuclear non-heme iron enzymes. These systems are described below.
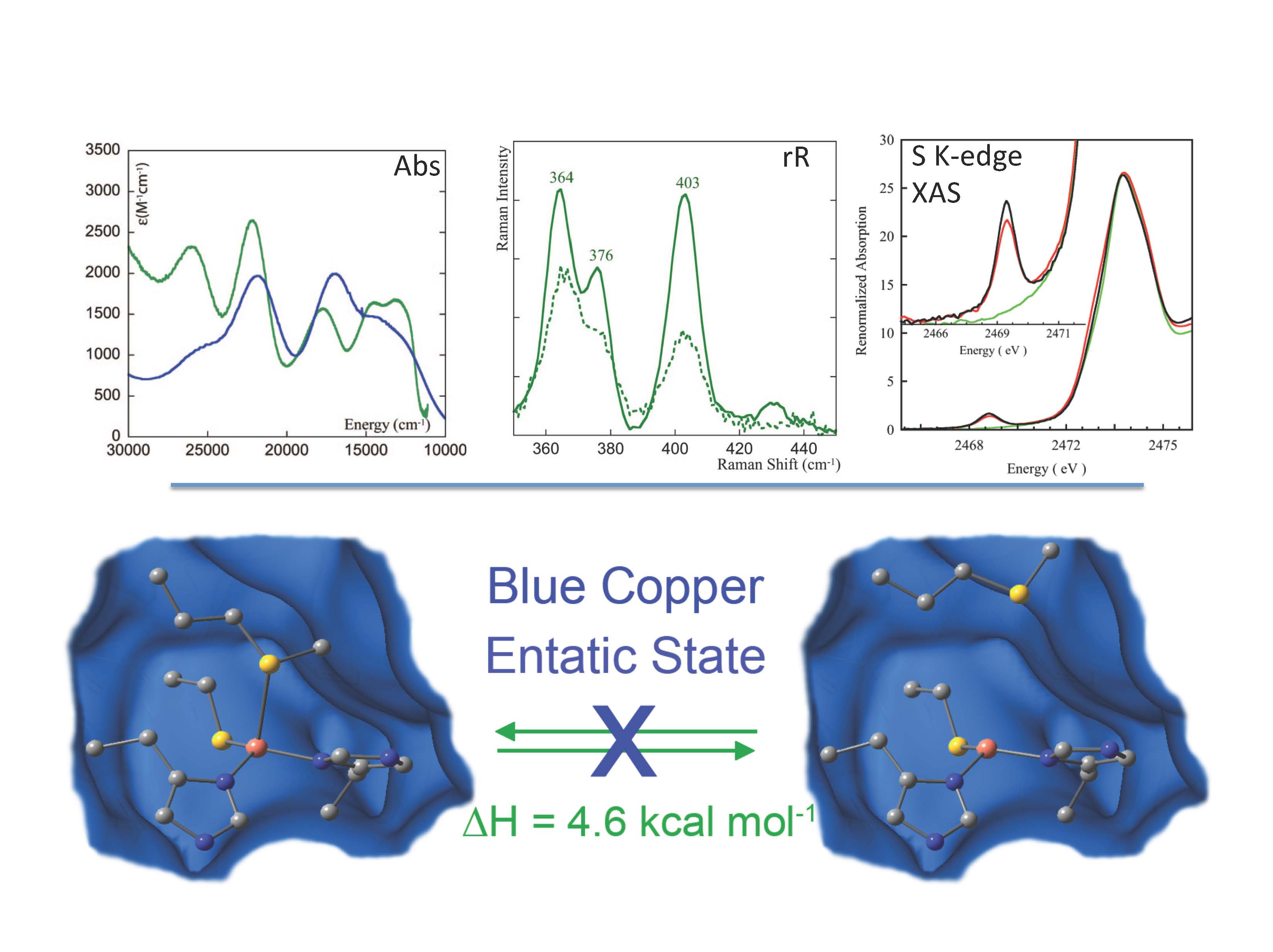
Metal sites involved in biological electron transfer (ET) are very diverse and include blue copper, CuA, the 1-, 2-, 3-, and 4-iron sulfur centers in rubredoxin, ferredoxins, and HiPIP, and the low-spin heme sites in cytochromes. Also relevant are heterometal-iron sulfur clusters and dithiolene-Mo oxo-transferases which are involved in multielectron small molecule activation, atom transfer, and proton coupled electron transfer (PCET). These active sites generally exhibit unusual spectroscopic features reflecting novel geometric and electronic structures that can make major contributions to function.
Our research group utilizes a wide-range of spectroscopies combined with density functional calculations to understand these electronic structures, the origin of their geometric structures, and possible contributions to function. The spectroscopies utilized include multifrequency EPR, absorption, circular dichroism, variable-temperature, variable-field magnetic circular dichroism (VTVH MCD), resonance Raman (rR) excitation profiles, variable energy photoelectron spectroscopy (VEPES), ligand K- and metal K- and L-edge X-ray absorption spectroscopies (XAS). These spectroscopic methods have been used to evaluate electronic structure calculations (ligand field and density functional) and the combination is being applied to obtain new insight into copper and iron sulfur ET proteins, iron-heme sites, and the non-innocent nature of the dithiolene ligand found in molybdoenzymes.
-
(1)“Recent Advances in Understanding Blue Copper Proteins” E. I. Solomon, R. G. Hadt, Coord. Chem. Rev. 2011, 255, 774. [DOI: 10.1016/j.ccr.2010.12.008]
-
(2)“Sulfur K-Edge X-ray Absorption Spectroscopy and Density Functional Calculations on Mo(IV) and Mo(VI)═O Bis-dithiolenes: Insights into the Mechanism of Oxo Transfer in DMSO Reductase and Related Functional Analogues” A. L. Tenderholt, J.-J. Wang, R. K. Szilagyi, R. H. Holm, K. O. Hodgson, B. Hedman, E. I. Solomon, J. Am. Chem. Soc. 2010, 132, 8359. [DOI: 10.1021/ja901369c]
-
(3)“Thermodynamic equilibrium between blue and green copper sites and the role of the protein in controlling function” S. Ghosh, X. Xie, A. Dey, Y. Sun, C. P. Scholes, E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 4969. [DOI: 10.1073/pnas.0900995106]
Copper Active Sites in Biology
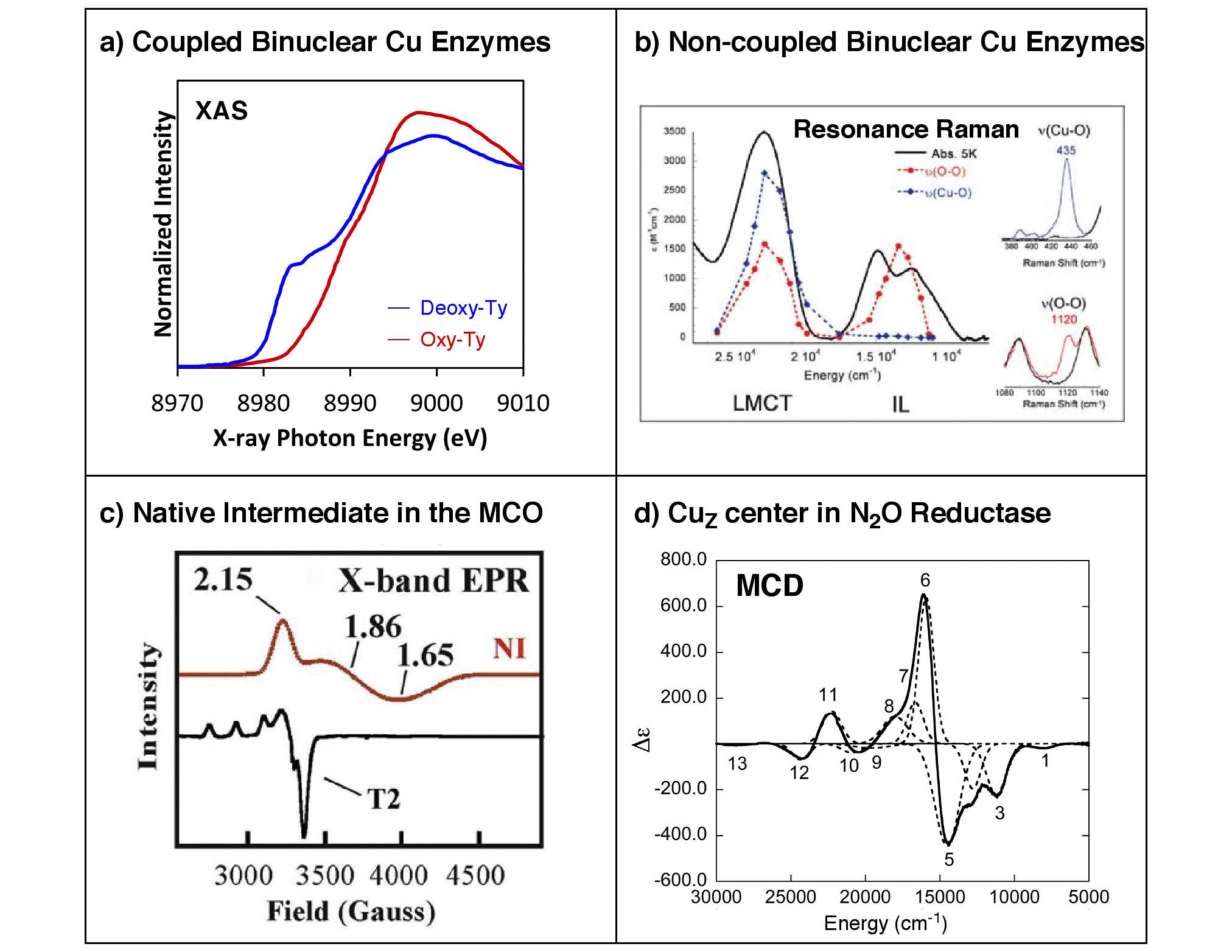
Copper proteins are involved in a wide range of biological oxidation-reduction processes. These include long-range electron transfer, dismutation of superoxide, reduction of nitrite and nitrous oxide, and reversible binding, transport, activation, and two- or four-electron reduction of dioxygen to peroxide or water that are coupled to substrate oxidation or proton pumping. This diversity can be attributed to the unique geometric and electronic structures of the copper active sites that are intricately tailored for their specific functions.
Biological copper active sites can be classified by their unique spectral features. These have been classically divided into type 1 “blue”, type 2 (T2) “normal”, and type 3 (T3) “coupled-binuclear” sites. However, recent studies on cytochrome c oxidase and nitrous oxide reductase have unveiled two novel classes of copper centers, the binuclear CuA and the tetranuclear CuZ sites, both of which have mixed-valent Stot = 1/2 ground states. In addition, different types of copper centers are found in clusters, as in the trinuclear copper cluster site of the multicopper oxidases that are comprised of a T2 and a T3 site. Interestingly, the trinuclear copper cluster, upon reacting with O2, produces a four-electron reduced oxygen intermediate that exhibits unusual spectral features that derive from the oxo-bridge mediated superexchange interactions among the three Cu(II) centers.
Our research covers a broad range of copper proteins, that mainly includes (a) coupled-binuclear Cu proteins involved in O2 binding/transport and activation (hemocyanin, tyrosinase, and catechol oxidase), (b) non-coupled binuclear Cu proteins involved in O2 activation and substrate hydroxylation (dopamine b-hydroxylase and peptidylglycine b-hydroxylating monooxygenase), (c) the multicopper oxidases involved in O2 reduction (laccase, Fet3p, CueO, CotA, Ascorbate Oxidase, and Ceruloplasmin), and (d) the CuZ active site in the nitrous oxide reductase involved in the N2O reduction. (T1 blue and CuA sites are mentioned in the ET section) Elucidation of the unique spectral and structural features of these active sites and their intermediates provides detailed insights into the nature of these sites that can be applied toward understanding their contributions to the unique catalytic functions in biology.
-
(1)“Spectroscopic Elucidation of a New Structure Type in Heme/Cu Dioxygen Chemistry: Implications for O–O Bond Rupture in Cytochrome c Oxidase” M. T. Kieber-Emmons, M. F. Qayyum, Y. Li, Z. Halime, K.O. Hodgson, B. Hedman, K. Karlin, E. I. Solomon, Angew. Chem. Intl. Ed. 2011, Published Online. [DOI: 10.1002/anie.201104080]
-
(2)“X-ray Absorption Spectroscopic and Computational Investigation of a Possible S···S Interaction in the [Cu3S2]3+ Core” R. Sarangi, L. Yang, S. G. Winikoff, L. Gagliardi, C. J. Cramer, W. B. Tolman, E. I. Solomon, J. Am. Chem. Soc. 2011, 133, 17180. [DOI: 10.1021/ja111323m]
-
(3)“Copper Dioxygen (Bio)inorganic Chemistry” E. I. Solomon, J. W. Ginsbach, D. E. Heppner, M. T. Kieber-Emmons, C. H. Kjaergaard, P. J. Smeets, L. Tian, J. S. Woertink, Faraday Disc. 2011, 148, 11. [DOI: 10.1039/c005500j]
-
(4)“Systematic Perturbation of the Trinuclear Copper Cluster in the Multicopper Oxidases: The Role of Active Site Asymmetry in its Reduction of O2 to H2O” A. J. Augustine, C. Kjaergaard, M. Qayyum, L. Ziegler, D. Kosman, K. O. Hodgson, B. Hedman, E. I. Solomon, J. Am. Chem. Soc. 2010, 132, 6057. [DOI: 10.1021/ja909143d]
-
(5)“Spectroscopic and Computational Studies of an End-on Bound Superoxo-Cu(II) Complex: Geometric and Electronic Factors that Determine the Ground State” J. S. Woertink, L. Tian, D. Maiti, H. R. Lucas, R. A. Himes, K. D. Karlin, F. Neese, C. Würtele, M. Holthausen, E. Bill, J. Sundermeyer, S. Schindler, E. I. Solomon, Inorg. Chem. 2010, 49, 9450. [DOI: 10.1021/ic101138u]
-
(6)“Geometric and Electronic Structure Differences Between the Type 3 Copper Sites of the Multicopper Oxidases and Hemocyanin/Tyrosinase” J. Yoon, S. Fujii, E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6585. [DOI: 10.1073/pnas.0902127106]
-
(7)“Reaction Coordinate of a Functional Model of Tyrosinase: Spectroscopic and Computational Characterization” B. T. Op’t Holt, M. A. Vance, L. M. Mirica, D. Heppner, T. D. P. Stack, E. I. Solomon, J. Am. Chem. Soc. 2009, 131, 6421. [DOI: 10.1021/ja807898h]
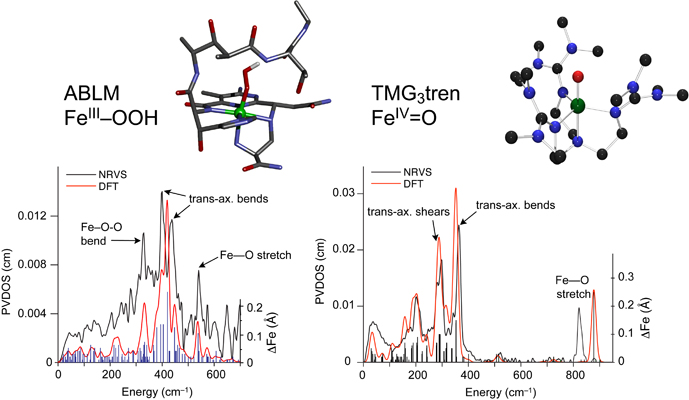
A large group of mononuclear non-heme iron enzymes bind and activate dioxygen to catalyze a variety of key biochemical transformations, including many of medical, pharmaceutical and environmental significance. These enzymes utilize high-spin FeII active sites and additional reducing equivalents from cofactors or substrates to react with O2 to yield iron-oxygen intermediates competent to transform substrate to product. While FeII sites have been difficult to study due to the lack of dominant spectroscopic features, a spectroscopic methodology has been developed which allows the elucidation of the geometric and electronic structures of these active sites and provides molecular level insight into the mechanisms of catalysis.
These studies provide key insight into the mechanisms of oxygen activation, active site features that contribute to differences in reactivity and, combined with theoretical calculations and model studies, the nature of oxygen intermediates active in catalysis.
-
(1) NRVS Studies of the Peroxide Shunt Intermediate in a Rieske Dioxygenase and Its Relation to the Native FeII O2 Reaction. Sutherlin, K.D.; Rivard, B.S.; Böttger, L.H.; Liu, L.V.; Rogers, M.S.; Srnec, M.; Park, K.; Yoda, Y.; Kitao, S.; Kobayashi, Y.; Saito, M.; Seto, M.; Hu, M.; Zhao, J.; Lipscomb, J.D. and Solomon, E.I., J. Am. Chem. Soc. 2018, 140, 5544. [DOI: 10.1021/jacs.8b01822]
-
(2) O2 Activation by Non-Heme Iron Enzymes. Solomon, E. I.; Goudarzi, S.; and Sutherlin, S. D. Biochemistry 2016, 55, 6363. [DOI: 10.1021/acs.biochem.6b00635]
-
(3) Nuclear Resonance Vibrational Spectroscopic Definition of Peroxy Intermediates in Nonheme Iron Sites. Sutherlin, K. D.; Liu, L. V.; Lee, Y-M.; Kwak, Y.; Yoda, Y.; Saito, M.; Kurokuzu, M.; Kobayashi, Y.; Seto, M.; Que, L., Jr.; Nam, W.; Solomon, E. I. J. Am. Chem. Soc. 2016, 138, 10294 [DOI: 10.1021/jacs.6b07227]
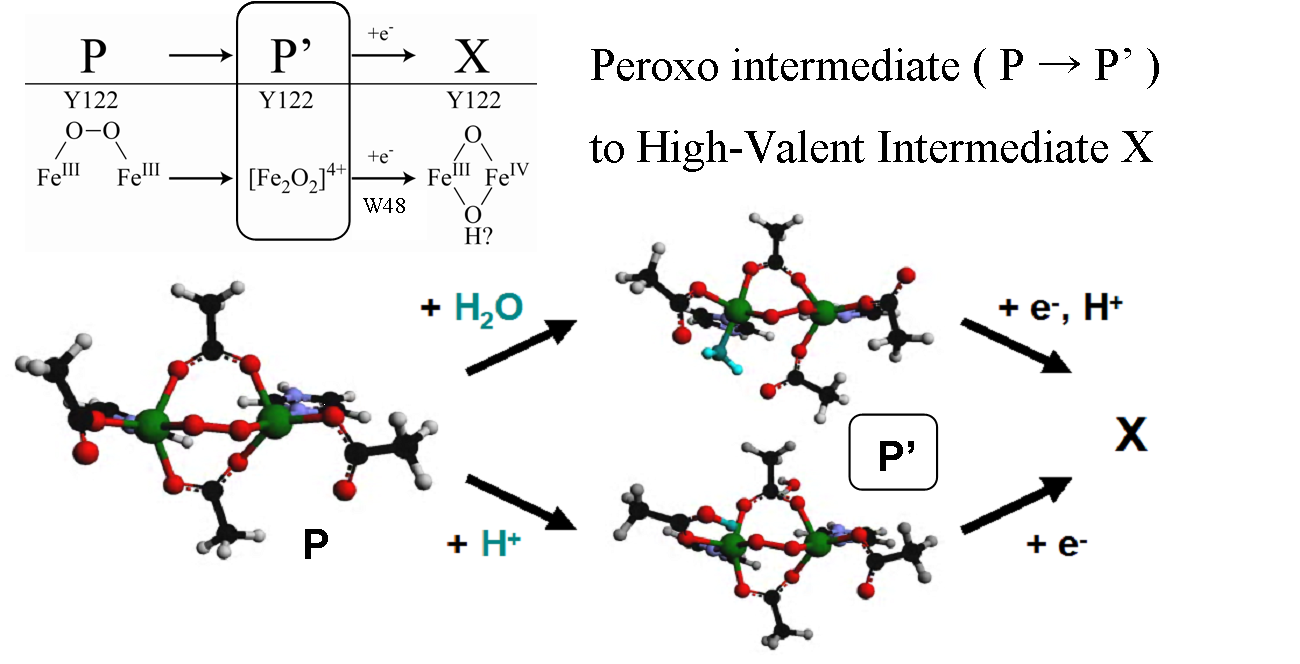
Binuclear non-heme iron proteins have been shown to carry out a variety of different reactions through activation of dioxygen. These include O2-binding (hemerythrin), activation for hydroxylation (methane monooxygenase, MMO), desaturation (Δ9 desaturase, Δ9D), tyrosine radical formation (ribonucleotide reductase, RR by a proton coupled electron transfer (PCET) process), reduction by two-electrons (ferroxidase site of ferritin, and the related peroxidase site of rubrerythrin), and its four-electron reduction to water (rubredoxin: O2 oxidoreductase, NO reductase, and the alternative oxidase in plants).
Despite a largely conserved ligand motif around the active sites in these enzymes, our studies utilizing absorption, resonance Raman, XAS, circular dichroism, magnetic circular dichroism (MCD), and variable-temperature, variable-field (VTVH) MCD spectroscopies applied in combination with density functional calculations provide significant insight into how geometric and electronic changes in the active site affect reactivity and allow for this diverse range of reactions to occur.
In addition to studying the resting states of these proteins, we are able to use rapid-freeze quench techniques to trap Fe2III-peroxide and high valent iron-oxo intermediates, as well as study other relevant oxygen-non-heme diiron model complexes and de novo designed protein analogues that have been synthesized. The spectroscopic data collected on these samples give great insight into the electronic structure and ligand environment of each Fe as well as the bridging between the irons. When combined with DFT calculations, we are able to obtain greater understanding of the structure/function correlations over the range of these binuclear non-heme iron enzymes and further elucidate their reactivity and reaction pathways.
-
(1) ”Peroxo-Type Intermediates in Class I Ribonucleotide Reductase and Related Binuclear non-Heme Iron Enzymes” K. Jensen, M. D. Clay, C. B. Bell III, E. I. Solomon, J. Am. Chem. Soc. 2009, 131, 12155. [DOI: 10.1021/ja809983g]
(2) “CD and MCD Spectroscopic Studies of the Two DPS Mini-Ferritin Proteins From B. Anthracis: Role of O2 and H2O2 Substrates in Reactivity of the Di-Iron Catalytic Centers” J. K. Schwartz, X. S. Liu, T. Tosha, A. R. Diebold, E. C. Theil, E. I. Solomon, Biochemistry 2010, 49, 10516. [DOI: 10.1021/bi101346c]
- Transition metal ion (TMI) containing zeolites offer a unique venue to study metal mediated small molecule activation (e.g., O2, NO, and N2O). The resulting MxOy species are often unprecedented due to the difficulty associated with their synthesis and isolation using traditional methods. Not only do they represent unique chemical structures in inorganic chemistry, but they often display intriguing reactivity. For example, activation of Fe- and Cu-ZSM-5 with N2O and O2 generates reactive intermediates capable of selectively oxidizing methane to methanol. Despite their intrinsic reactivity, these species can be stabilized for detailed spectroscopic analysis. We currently employ diffuse reflectance, resonance Raman (rR), electron paramagnetic resonance (EPR), and other spectroscopies to probe the geometric and electronic structures of the reactive intermediates in zeolites. When combined with density functional theory (DFT) calculations, these data provide significant links between structure and reactivity. These concepts are then used to provide an insightful correspondence between heterogeneous and biological catalysis.
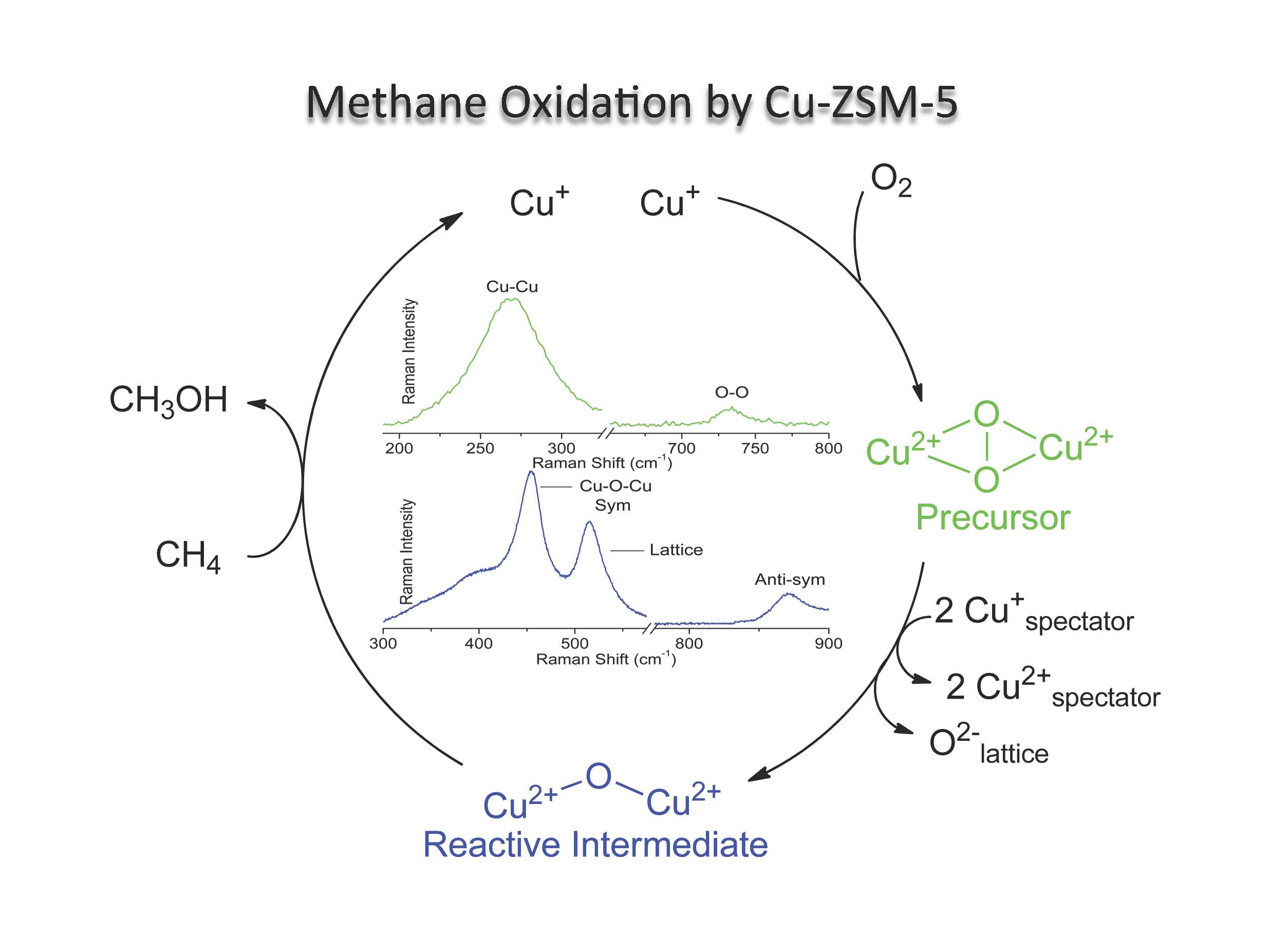
-
(1)Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes. Snyder, B. E. R.; Bols, M. L.; Schoonheydt, R. A.; Sels, B.F.; and Solomon, E. I., Chem. Rev. 2018, 118, 2718. [DOI: 10.1021/acs.chemrev.7b00344]
(2)Structural characterization of a non-heme iron active site in zeolites that hydroxylates methane. Snyder, B. E. R.; Böttger, L. H.; Bols, M.L., Yan, J.J.; Rhoda, H.M.; Jacobs, A.B.; Hu, M.Y.; Zhao, J.; Alp, E.E.; Hedman, B.; Hodgson, K.O.; Schoonheydt, R.A.; Sels, B.F. and Solomon, E.I., PNAS 2018, 115, 4565. [DOI: 10.1073/pnas.1721717115]
-
(1)Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes. Snyder, B. E. R.; Bols, M. L.; Schoonheydt, R. A.; Sels, B.F.; and Solomon, E. I., Chem. Rev. 2018, 118, 2718. [DOI: 10.1021/acs.chemrev.7b00344]
