The Voss group develops methods for an accurate computational description of surface chemistry. These developments consist of devising semi-empirical exchange-correlation functionals for density functional theory (DFT) and algorithmic improvements for catalytic rate predictions based on micro-kinetic modeling.
The group develops exchange-correlation (XC) functionals by means of fitting the functional form against benchmark data. The benchmark data consists of both higher level of theory (than DFT, i.e. wave function methods for molecules) and experimental data (for bulk cohesive and elastic properties, surface reactions, etc.) [Brown, Maimaiti, Trepte, Bligaard, and Voss, J. Comput. Chem. 42, 2004 (2021)].
×
![]()
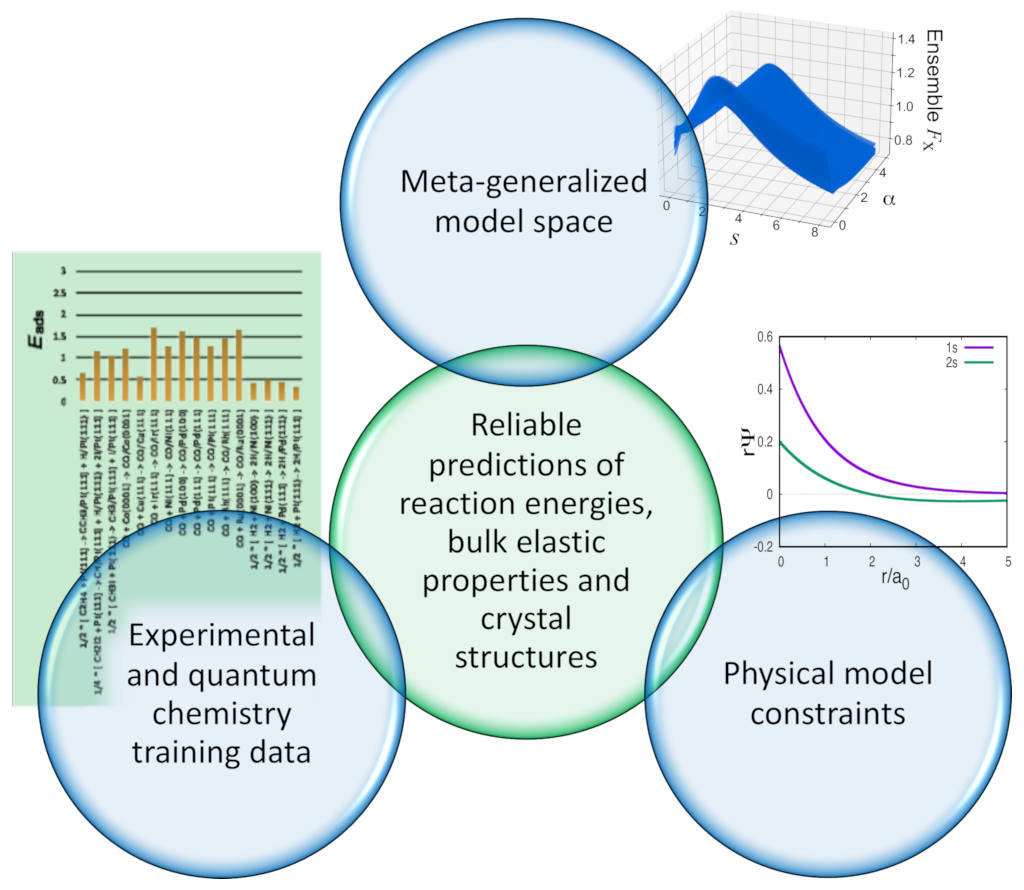
Schematic of MCML training approach: experimental and quantum chemistry benchmark data for reaction energetics and bulk cohesive and lattice properties are combined with physical constraints in a meta-generalized model space.
Combining these training data with physical model constraints allows for the optimization of the XC functional towards a transferable functional with improved accuracy for surface and gas phase reaction energies without sacrificing the good description of crystal structures with existing approaches:
×
![]()
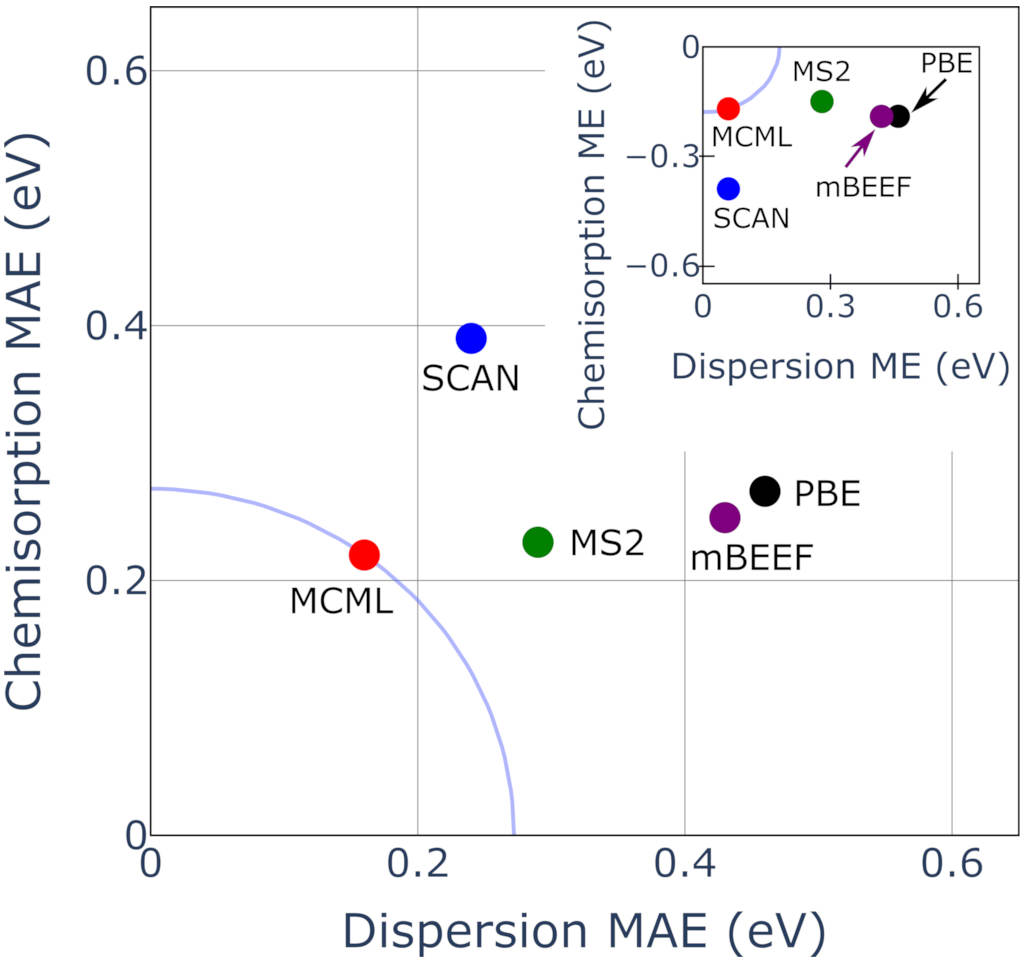
Mean absolute and signed errors for XC functional performance on predicting adsorption energies in the ADS41 [
Mallikarjun Sharada, Karlsson, Maimaiti, Voss, Bligaard, Phys. Rev. B 100, 035439 (2019)] dataset. From
Brown, Maimaiti, Trepte, Bligaard, and Voss, J. Comput. Chem. 42, 2004 (2021). Copyright (2021) Wiley Periodicals LLC.
The resulting multi-purpose, constrained and machine-learned (MCML) functional is available in libxc version 5.1.6 and above.
Using Bayesian statistics, the fitted functionals allow for an inferred estimate of the uncertainty in the computed reaction energetic predictions. For more details on the functional development work see https://www.slac.stanford.edu/~vossj/project/xc-functionals/.
×
![]()
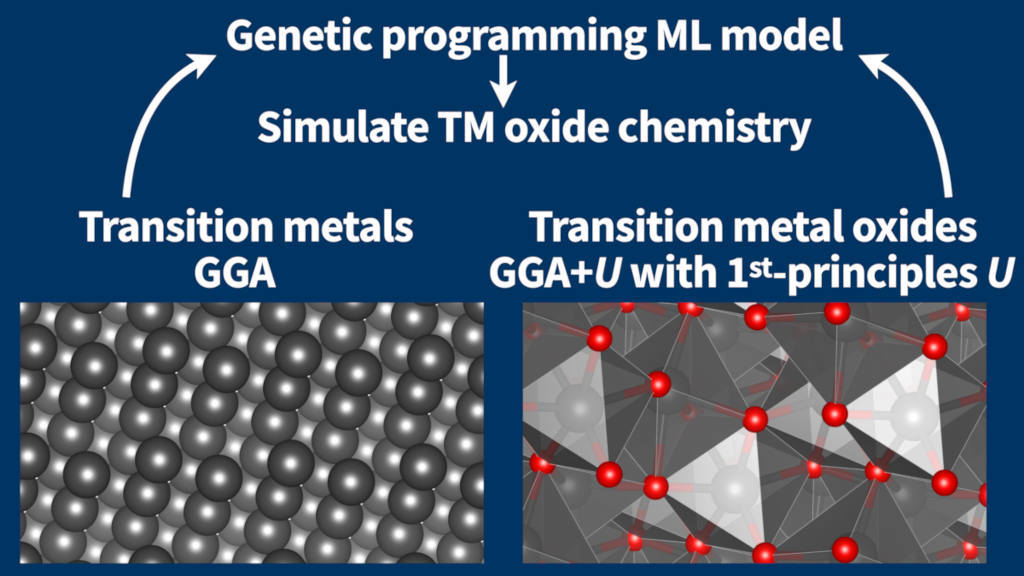
Overview of machine-learning model for prediction of transition metal oxide heat of formation through a combination of GGA and GGA+U simulations with Hubbard U-parameters computed from first principles.
When self-interaction errors are significant due to strong Coulomb interactions, e.g., in the case of transition metal oxides, approaches beyond semi-local DFT are necessary to qualitatively describe the electronic structure of these strongly correlated systems correctly. Here, we have employed the machine-learning technique of genetic programming to devise a model that enables the prediction of transition metal oxide heat of formation and other reaction energies involving both localized and delocalized d-electrons from a combination of GGA and GGA+U simulations with first-principles Hubbard U-parameters [Voss, J. Phys. Commun. 6, 035009 (2022)].
×
![]()
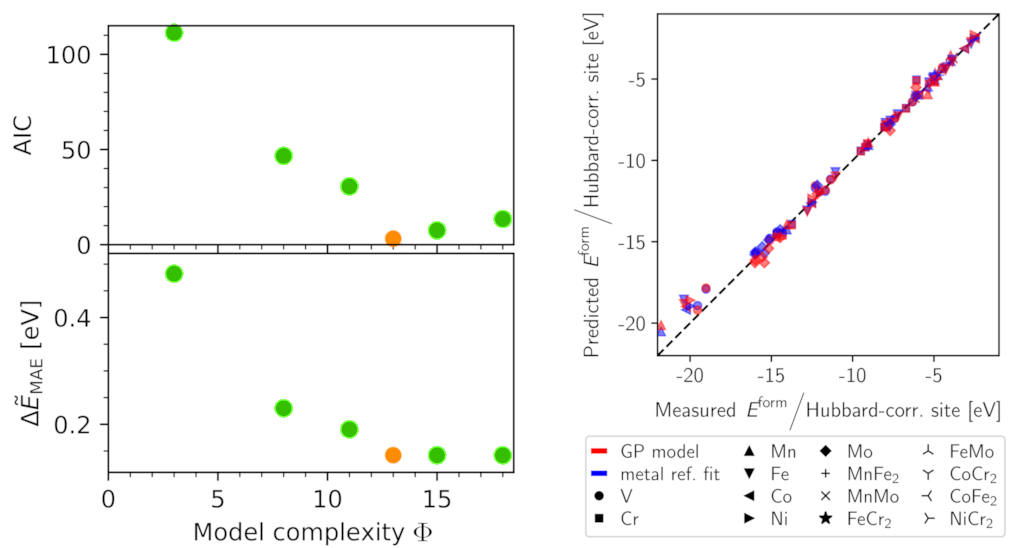
The genetic programming search led to a simple model that does not explicitly depend on the ionic types at the Hubbard-corrected sites and thus allows for computation of transition metal oxide reaction energies in the absence of experimental reference data to fit corrections required in previous approaches.
×
![]()
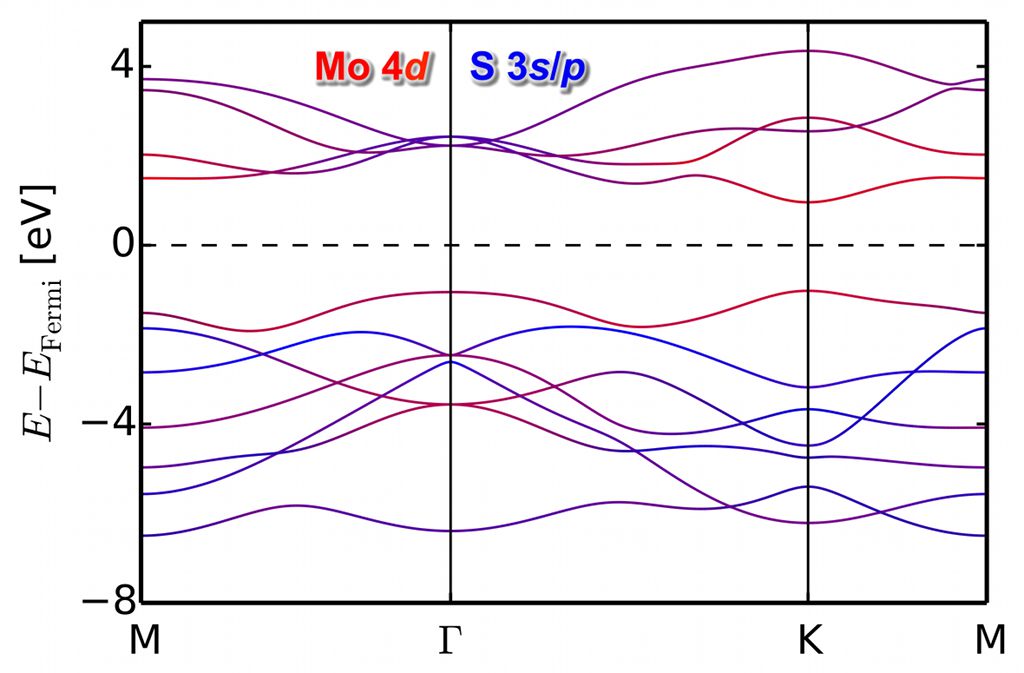
GLLBsc band structure of monolayer MoS2. The DFT band gap has been augmented according to the derivative-discontinuity correction of the GLLBsc formalism. The predicted direct band gap at $\textrm{K}$ of about 2eV agrees well with experiment (exciton binding energy corrections have been neglected). Red color indicates dominant Mo $4d$ and blue S $3s$/$p$ character.
The group also works on ab initio functionals for efficient predictions of spectral properties. In these typically orbital dependent functionals, corrections to the inherent band gap problems of DFT are computed from first principles. More information can be found here: https://www.slac.stanford.edu/~vossj/project/perovskite-lightabsorbers/.
×
![]()
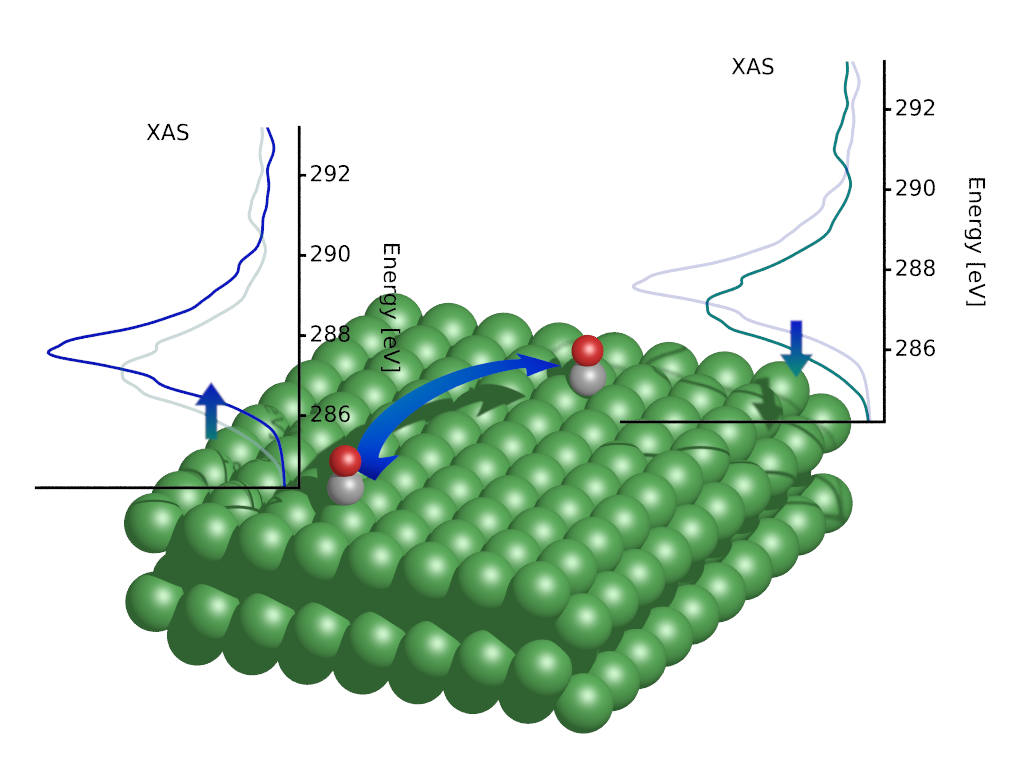
To gain a deeper understanding into catalytic selectivity and intermediate states during heterogeneous catalytic reactions, the group performs X-ray spectroscopical simulations to guide analysis of ultrafast catalysis experiments using free electron lasers. More information can be found here: https://www.slac.stanford.edu/~vossj/project/ultrafast-catalysis/.




