PRIONS -- Beware what you eat!
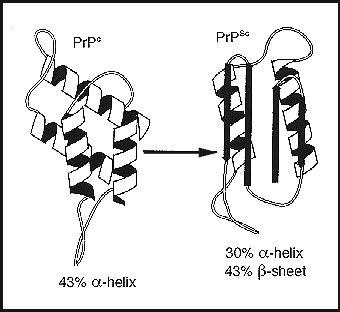

Image 1 taken from:http://www.agric.gov.ab.ca/images/surveillance/prions.gif
Image 2 taken from:http://www.le.ac.uk/biology/research/phyto/prions.jpg
Prions, or infectious proteins, received a lot of media attention this year due to the revelation that it had somehow reached the United States. Although much was made of this, the focus of this page will be to look past the animal prion diseases and look more at how they affect humans.
Prion diseases
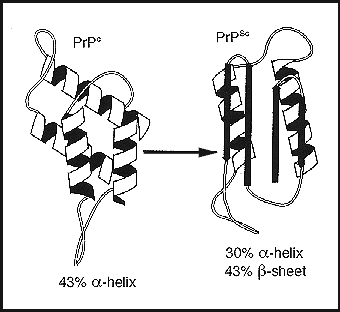
Prions are the primary cause of Transmissible Spongiform Encephalophathies (TSEs). They occur in both humans and other animals.
Animal forms: Scrapie,BSE, Transmissible mink encephalopathy (TME), chronic wasting disease, Feline spongiform encephalopathy (FSE)(for more information on these forms please visit the website of John B. Mumm, Humans and Viruses, 1999 at http://www.stanford.edu/group/virus/prion/prion2.html )
Human forms: kuru, Creutzfeldt-Jakob disease (CJD), Gerstmann-Straussler syndrome (GSS), fatal familial insomnia (FFI)
All the TSEs are characterized by neuronal loss, accompanied by amyloid plaques or fibril formation after spongiform degeneration of brain tissue. They are progressive neuronal diseases that are always fatal.
Symptoms: Dementia, ataxia (loss of muscle control of voluntary movements) from the loss of brain function due to degeneration.
Outcome: Once symptoms appear, death results in 6 months -1 year. There is no cure and our own bodies can't even mount an immune response to this pathogen.
Epidemiology
Pretty much all of the human TSEs occur throughout the population at an equal frequency, but Kuru was endemic among the Fore People of New Guinea until the ritualistic practice of cannibalism was ended there. (Look at to get a full look at the history of prion disesases) Also CJD tends to occur in older populations with very few cases occuring in individuals under the age of 40.
Transmission: Inoculation or ingestion of brain tissue, sporadic mutation, or it can be inherited.
Intubation period: This can be as long as 40 years as in Kuru, although it appears to be much shorter for viariant CJD cases.
GSS, FFI, and CJD are inheritable. This kind of transmission occurs when the dominant gene associated with the mutation in the prion protein is inherited. When that gene is present, there is close to a 100% likelihood that progression to one of these TSEs will result.
CJD can also occur both sporadically (at about a frequency of 1/million) or via inoculation. No matter how transmitted, once the mutation has occured in the protein to result in the prion, it can be transmitted as an infectious disease.
Prevention and Treatment
Presently there is no real treatment for any of the TSEs. Prions are very hardy and almost indestructible. Disinfection must occur with agents like environ LpH. Also, all efforts to keep out of contact with contaminated tissues should be made. More information on protocol can be found at the following websites:
http://www.cjdsurveillance.com/
This article tells of possible use of antibodies for treating prion diseases http://www.nature.com/nsu/030303/030303-7.html
This link shows the protocol for Johns Hopkins http://www.hopkins-heic.org/pdf/ifc032.pdf
http://www1.umn.edu/eoh/hazards/hazardssite/prions/prioncontrol.html
Image taken from http://www1.umn.edu/eoh/hazards/hazardssite/prions/prioncontrol.html
Update 2004
- Fatal familial isomnia has been found for the first time in a family of Chinese descent!
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}