
New Therapies and Drugs
Bryostatin and the Bryologs
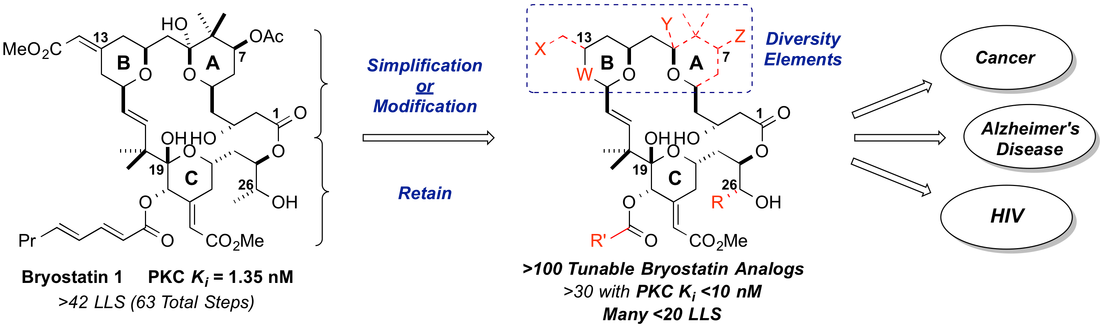
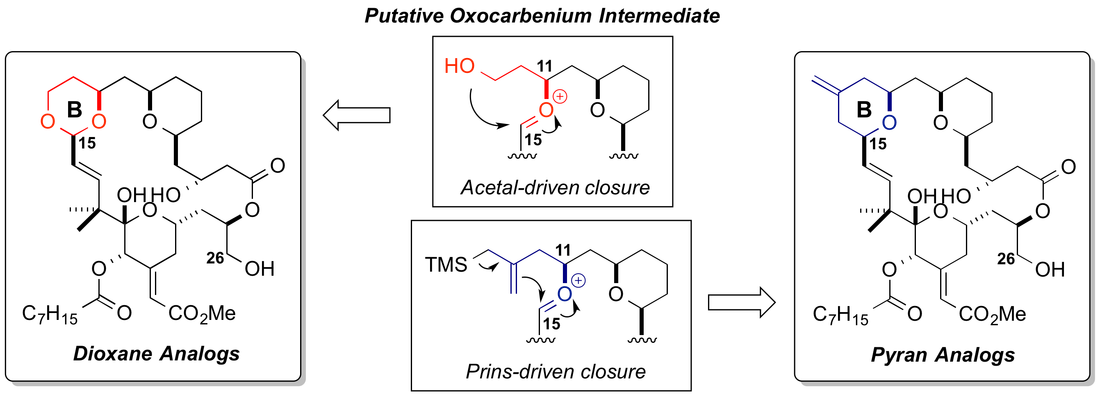
That bryostatin 1 has already been tested in human trials increases its appeal as a target and underscores the need for a reliable supply of a more effective agent. In fact, patient accrual in a recent National Cancer Institute clinical trial with bryostatin 1 was terminated given the awareness of the clinical investigators of more potent bryostatin analogs in development. Research on and clinical studies of bryostatin has been impeded by its low natural abundance. Isolated yields from natural sources are low (10-3 to 10-8 %) and variable; the good manufacturing practices (GMP) production required 14 tons of the marine bryozoan Bugula neritina to provide just 18 grams of bryostatin 1. While this supply was sufficient to initiate preclinical and clinical research, environmental and economic factors have severely limited further development of the source organism and its aquaculture production. Furthermore, while a superb lead, bryostatin suffers from off-target toxicities that have been revealed in clinical studies.
Gnidimacrin

Daphnetoxins


Prostratin
Prostratin is a naturally occurring tigliane and represents the active constituent of a traditional tonic used by Samoan healers to treat a local type of hepatitis. After its identification through an NIH effort to discover novel medicinal leads, prostratin was shown to stimulate the proliferation of HIV in latently infected cells. Unfortunately, the current isolation process is low yielding (0.005%) and at the moment does not appear to provide the required quantities for therapeutic advancement of prostratin. More importantly, we could in principle design more effective agents.
We have established a program that addresses the prostratin supply problem, has produced the first synthetic source of prostratin and is directed at the optimization of the therapeutic properties of prostratin through function oriented analog design and synthesis. We have shown that prostratin can be generated through a concise synthesis starting with phorbol (5 steps, ~16% overall yield), which can be obtained in quantity either from commercial sources or directly through isolation from croton oil. More importantly, we have now produced agents that are significantly more potent than prostratin.

References
- DeChristopher, B.A.; Loy, B.A.; Marsden, M.D.; Schrier, A.J.; Zack, J.A.; Wender, P.A. “Designed, synthetically accessibility bryostatin analogues potently induce activation of latent HIV reservoirs in vitro.” Nature Chem., 2012, 4, 705-710.
- Wender, P.A.; Billingsley, K.L. “Lead Diversification through a Prins-Driven Macrocyclization Strategy: Application to C13-Diversified Bryostatin Analogues.” Synthesis, 2013, 45 (13), 1815-1824.
- Ogawa, Y.; Painter, P.P.; Tantillo, D.J.; Wender, P.A. "Mechanistic and Computational Studies of Exocyclic Stereocontrol in the Synthesis of Bryostatin-like Cis-2, 6-Disubstituted 4-Alkylidenetetrahydropyrans by Prins Cyclization." J. Org. Chem., 2013, 78(1), 104-115.
- DeChristopher, B. A.; Fan, A. C.; Felsher, D. W.; Wender, P. A. “ ‘Picolog,’ a synthetically-accessible bryostatin analog, inhibits growth of MYC-induced lymphoma in vivo.” Oncotarget, 2012, 3, 58-66.
- Wender, P. A.; Baryza, J. L.; Brenner, S. E.; DeChristopher, B. A.; Loy, B. A.; Schrier, A. J. “Design, synthesis and evaluation of potent bryostatin analogs that modulate PKC translocation selectivity.” Proc. Natl. Acad. Sci. USA, 2011, 108, 6721-6726.
- Wender, P. A.; Schrier, A. J. “Total Synthesis of Bryostatin 9.” J. Am. Chem. Soc., 2011, 133, 9228-9231.
- Wender, P. A.; Loy, B. A.; Schrier, A. J. “Translating nature’s library: the bryostatins and function-oriented synthesis.” Isr. J. Chem., 2011, 51, 453-472.
- Wender, P. A.; DeChristopher, B. A.; Schrier, A. J. “Efficient Synthetic Access to a New Family of Highly Potent Bryostatin Analogues via a Prins-Driven Macrocyclization Strategy,” J. Am. Chem. Soc., 2008, 6658.
- Wender, P. A.; Baryza, J.; Bennett, C.; Bi, C.; Brenner, S. E.; Clarke, M.; Horan, J.; Kan, C.; Lacote, E.; Lippa, Nell, P.; Turner, T.“The Practical Synthesis of a Novel and Highly Potent Analog of Bryostatin” J. Am. Chem. Soc., 2002, 124, 13648.
Gnidimacrin and Daphnetoxins Lead References:
- Wender, P.A.; Buschmann, N.; Cardin, N.B.; Jones, L.R.; Kan, C.; Kee, J-M.; Kowalski, J.A.; Longcore, K.E. “Gateway Synthesis of daphnane congeners and their protein kinase C affinities and cell-growth activities” Nature Chem. 2011, 3, 615.
- Wender, P. A.; D'Angelo, N.; Elitzin, V. I.; Ernst, M.; Jackson-Ugueto, E. E.; Kowalski, J. A.; McKendry, S.; Rehfeuter, M.; Sun, R.; Voigtlaender, D. “Function-Oriented Synthesis: Studies Aimed at the Synthesis and Mode of Action of 1-Alkyldaphnane Analogues ” Org. Lett. 2007, 1829.
- Wender, Bi, Buschmann, Gosselin, Kan, Kee, Ohmura, "Studies on Oxidopyrylium [5+2] Cycloadditions: Toward a General Synthetic Route to the C12-Hydroxy Daphnetoxins" Org. Lett. 2006, 8, 5373.
- Wender, Jesudason, Nakahira, Tamura, Tebbe, Ueno "The First Synthesis of a Daphnane Diterpene: The Enantiocontrolled Total Synthesis of (+)-Resiniferatoxin" J. Am. Chem. Soc. 1997, 119, 12976.
- Wender, Rice, Schnute, "The First Formal Asymmetric Synthesis of Phorbol" J. Am. Chem. Soc. 1997, 7611-7612.
Prostratin Lead Reference:
- Beans, E.J.; Fournogerakis, D.; Gauntlett, C.; Heumann, L.V.; Kramer, R.; Marsden, M.D.; Murray, D.; Chun, T.-W.; Zack, J.A.; Wender, P.A. “Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo.” Proc. Natl. Acad. Sci., USA, 2013, 110 (29), 11698-11703.
- Wender, P.A.; Kee, J.-M.; Warrington, J.M. “Practical Synthesis of Prostratin, DPP, and Their Analogs, Adjuvant Leads Against Latent HIV.” Science, 2008, 320 (5876), 649-652.