


Next:Registering.Up:Documentation
for structure software:Previous:ContentsContents
Introduction
The program structure implements a model-based clustering method
for inferring population structure using genotype data consisting of unlinked
markers. The methods used are introduced in a paper by Pritchard, Stephens
and Donnelly (2000), which is the appropriate citation for this program4.
Applications of our method include demonstrating the presence of population
structure, assigning individuals to populations, and identifying migrants
and admixed individuals. Briefly, we assume a model in which there are 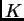 populations (where
may be unknown), each of which is characterized by a set of allele frequencies
at each locus. Individuals in the sample are assigned (probabilistically)
to populations, or jointly to two or more populations if their genotypes
indicate that they are admixed. It is assumed that within populations,
the loci are at Hardy-Weinberg equilibrium, and linkage equilibrium. Loosely
speaking, individuals are assigned to populations in such a way as to achieve
this. Our model does not assume a particular mutation process, and it can
be applied to most of the commonly used genetic markers including microsatellites,
SNPs and RFLPs, provided that they are unlinked (or at least, not so tightly
linked that they are in linkage-disequilibrium; see section 1.2).
While the computational approaches implemented here are fairly powerful,
some care is needed in running the program in order to ensure sensible
answers. For example, it is not possible to determine suitable run-lengths
theoretically, and this requires some experimentation on the part of the
user. The user should consult the accompanying paper (Pritchard
et al., 2000a) for a description of the uses and limitations of this
method. This document describes the use of this software, and supplements
the
(Pritchard
et al., 2000a) paper.
populations (where
may be unknown), each of which is characterized by a set of allele frequencies
at each locus. Individuals in the sample are assigned (probabilistically)
to populations, or jointly to two or more populations if their genotypes
indicate that they are admixed. It is assumed that within populations,
the loci are at Hardy-Weinberg equilibrium, and linkage equilibrium. Loosely
speaking, individuals are assigned to populations in such a way as to achieve
this. Our model does not assume a particular mutation process, and it can
be applied to most of the commonly used genetic markers including microsatellites,
SNPs and RFLPs, provided that they are unlinked (or at least, not so tightly
linked that they are in linkage-disequilibrium; see section 1.2).
While the computational approaches implemented here are fairly powerful,
some care is needed in running the program in order to ensure sensible
answers. For example, it is not possible to determine suitable run-lengths
theoretically, and this requires some experimentation on the part of the
user. The user should consult the accompanying paper (Pritchard
et al., 2000a) for a description of the uses and limitations of this
method. This document describes the use of this software, and supplements
the
(Pritchard
et al., 2000a) paper.
Subsections
Next:Registering.Up:Documentation
for structure software:Previous:ContentsContents
William Wen 2002-07-18