Manual
- Installation
- Using LSC
- Module descriptions
- Input files
- Output files
- Short read - Long read aligner
- Short-read coverage depth (SCD)
- Execution Time
 Installation
Installation
No explicit installation is required for LSC. You may copy the LSC binaries to any location as long as all the binaries (including Novoalign) are in the same directory or path.
But you need to Python2.6 installed in your computer. The modules "numpy" and "scipy" are also required. Please see LSC requirements for more details
Using LSC
Firstly, see the tutorial on how to use LSC on some example data.
In order to use LSC on your own data:
- Create an empty directory, this will be the working directory.
- Copy "run.cfg" from the LSC package to the working directory.
- Edit run.cfg to include paths to your python binary directory, data files and the paths of the temp folder and the output folder. You may also want to configure other parameters default settings. LSC uses external aligners to find short reads to long reads mappings. Latest LSC version supports BWA, Bowtie2, Novoalign and RazerS (v3) aligners. The aligners and their setting options could be modified through run.cfg file. The path to selected aligner should be set in your PATH env variable.
- Execute "/home/user/LSC_path/runLSC.py run.cfg" while in your working directory. or Execute "runLSC.py run.cfg, if all LSC executable files are in the default bin
- After a certain time execution will conclude. You can find results in the "output" directory.
Module: runLSC
"runLSC.py" is the main program in the LSC package. It calls other modules to run the full error correction on your data. Output is written to the "output" folder. Details of the output files are described in file formats. Its options are described in run.cfg file. You just need to run "runLSC.py" with a configuration file "run.cfg":
/home/user/LSC_path/runLSC.py run.cfgor If you have put all LSC executable files in the default path:
runLSC.py run.cfgIn vesion 3.0 or later, you can run LSC in two steps using 'mode' option in run.cfg. If for a reason LSC was terminated in correction step, you can restart the correction step without re-running the LR-SR mapping step.
- mode 0 (default): end-to-end LSC run.
- mode 1: generates compressed SR to LR alignment file (SR.fa.cps.nav) which is used in LSC correctiopn step.
- mode 2: runs LSC correction step. Note: make sure SR.fa.cps.nav file is already generated in temp folder. At this stage, LSC also uses some other intermediate files in temp folder which are generated after mode 0/1 run
To check LSC version: runLSC.py -v
To clean up temp folderpath (if you used "cleap_up=0" option): runLSC.py run.cfg -clean_upNote: temp_path, Nthreads1 and Nthreads2 from run.cfg file are used for removing intermediate files.
Input files
LSC accepts one long-read sequences file (to be corrected) and one short-read sequences file as input. The input files could be in standard fasta or fastq formats.
Note: As part of LSC algorithm, it generates homopolyer-compressed short-read sequences before alignment. If you have already run LSC with the same SR dataeset you can skip this step by using previously generated
homopolyer-compressed SR files. (You can find SR.fa.cps and SR.fa.idx in temp folderpath.)
The file locations and their format should be set in run.cfg file through pathfilename and filetype options.
Output files
There are four output files: corrected_LR.fa, corrected_LR.fq, full_LR.fa, uncorrected_LR.fa in output folder:
- corrected_LR
- As long as there are short reads (SR) mapped to a long read, this long read can be corrected at the SR-covered regions. (Please see more details from the paper).
The sequence from the left-most SR-covered base to the right-most SR-covered base is outputted in the file corrected_LR. The output readname format is
<original readname>|<percentage of corrected output sequence covered by short reads> example: m111006_202713_42141_c100202382555500000315044810141104_s1_p0/16|0.81
- full_LR
- Although the terminus sequences are uncorrected, they are concatenated with their corrected sequence (corrected_LR) to be a "full" sequence. Thus, this sequence covers the equivalent length as the raw read and is outputted in the file full_LR.fa
- uncorrected_LR
- This is the negative control. uncorrected_LR.fa contains the left-most SR-covered base to the right-most SR-covered base (equivalent region in corrected_LR) but not error corrected. Thus, it is fragments of the raw reads.
| SRs Coverage | Error Probability* |
| 0 | 0.275 |
| 1 | 0.086 |
| 2 | 0.063 |
| 3 | 0.051 |
| 4 | 0.041 |
| 5 | 0.034 |
| 6 | 0.028 |
| 7 | 0.023 |
| 8 | 0.018 |
| 9 | 0.014 |
| 10 | 0.011 |
| 11 | 0.008 |
| 12 | 0.005 |
| 13 | 0.002 |
| >= 14 | ~0.000 |
Module: filter_corrected_reads.py
exapmle: python bin/filter_corrected_reads.py 0.5 output/corrected_LR.fa > output/corrected_LR.filtered.fa
You can also select "best" reads for your downstream analysis by mapping corrected LRs to the reference genome or annotation (for RNA-seq analysis). Then, filter the reads by mapping score or percentage of base match (e.g. "identity" in BLAT)
Short read-Long read Aligner
LSC uses a short read aligner in the first step. By default, Bowtie2 is used. You can have BWA, , Novoalign or RazerS (v3) to run this step as well.
Default aligners setting are:
Bowtie2 : -a -f -L 15 --mp 1,1 --np 1 --rdg 0,1 --rfg 0,1 --score-min L,0,-0.08 --end-to-end --no-unal
BWA : -n 0.08 -o 10 -e 3 -d 0 -i 0 -M 1 -O 0 -E 1 -N
Novoalign* : -r All -F FA -n 300 -o sam
RazerS3 : -i 92 -mr 0 -of sam
You can change these settings through .cfg file. Please refer to their manuals for more details.
* Note: novoalign has limitation on read length. If you are using LSC with novoalign, please make sure your short reads length do not exceed maximum threashold.
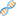
Following figures compare LSC correction results configured with different supported aligners. Identity metric is defined as number-of-matchs/error-corrected-read-length after aligning reads to reference genome using Blat.
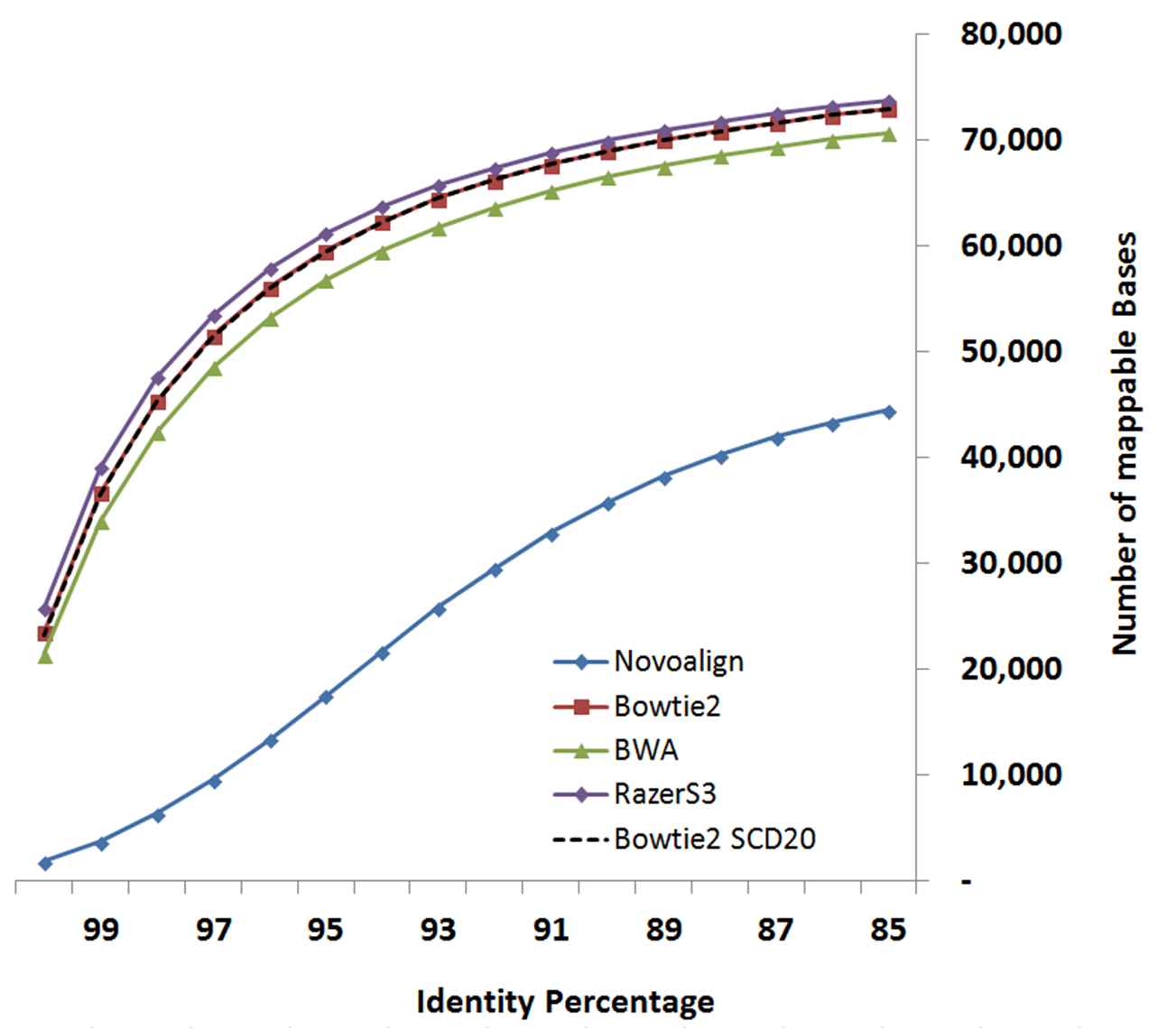
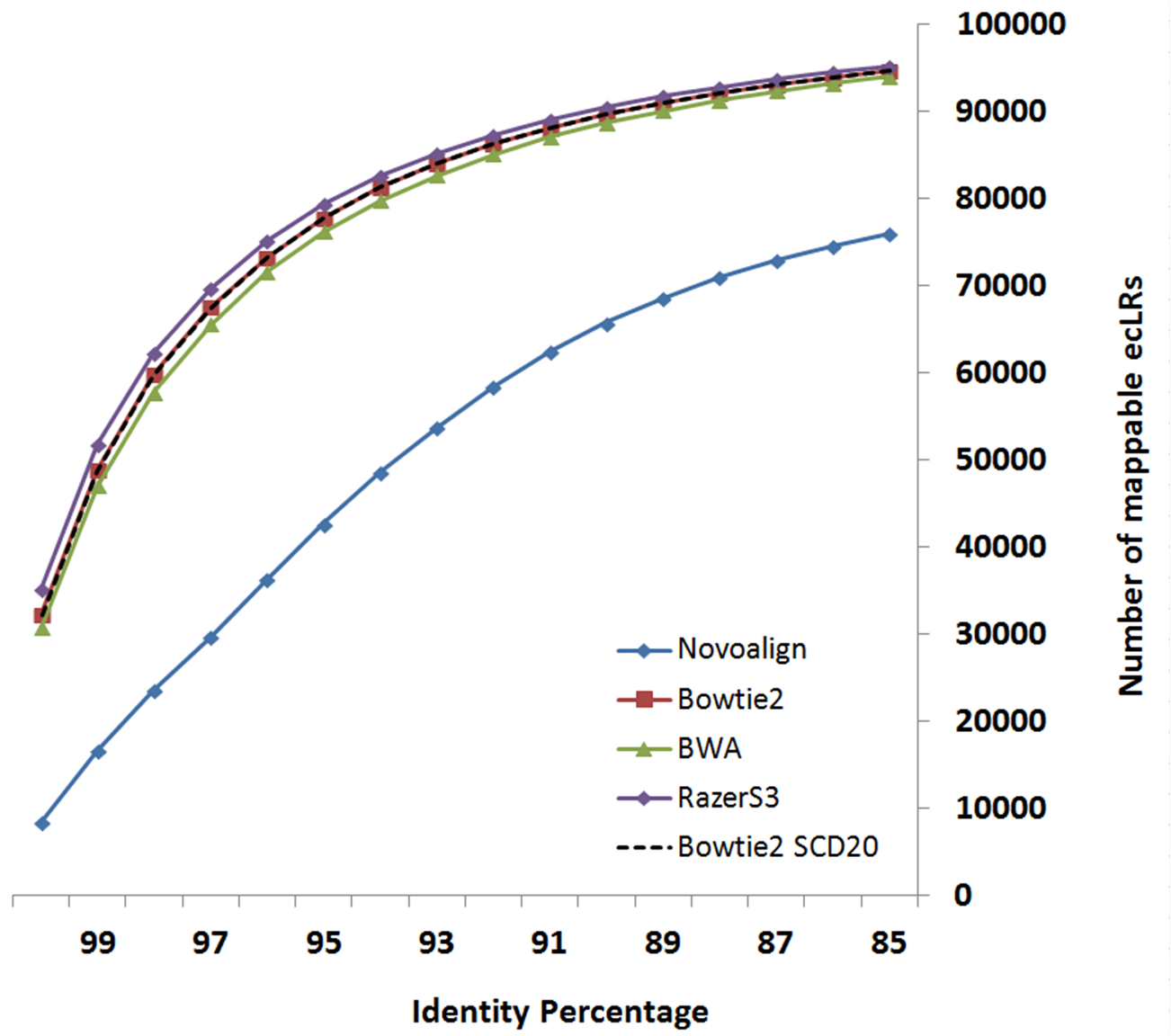
Data-set:
- LongReads: human brain cerebellum polyA RNA processed to enrich for full-length cDNA for
the PacBio RS platform under C2 chemistry conditions as CLR data
link
- ShortReads: human brain data from Illumina’s Human Body Map 2.0 project (GSE30611)
Based on your system configuration, you can select the aligner which fits better with your CPU or Memory resources.
The below table is derived experimentally by running LSC using different aligners on above-mentioned data-set.
| CPU | Memory | |
| BWA | Less | Less |
| Bowtie2 | More | Less |
| RazerS3 | More | More |
Short-read coverage depth (SCD)
LSC uses consensus of short-read mapping results to correct long read sequences. In case of having high SR coverage, pile of SRs mapped to a LR segment would significantly increase running time and memory usage in correction step, while having repetitive (redundant) information. By setting SCD parameter in run.cfg file, LSC uses a probabilistic algorithm to randomly select bounded number of SR alignemt results for each LR region in order to maintain expected SR coverage depth of SCD value. This would eliminate high memory peaks in corection step due to pile of SRs mapped in high coverage or repetitive regions. Based on our experiment on multilpe datasets, setting SCD = 20 gave comparable results w.r.t SCD = -1 (using all alignment results,i.e. without any bounded coverage limit).
Execution Time
Following CPU and execution times are suggested-usage using LSC.0.2.2 and LSC 1.alpha on our clusters with six thread. These figures will greatly differ based on your system configuration.
100,000 PacBio long reads X 64 million 75bp Illumina short reads (Dataset)
- LSC 1.alpha (w/ bowtie2, SCD=20): Time = 09:40:33, Max vmem = 8.532G
- LSC.0.2.2(w/ novoalign): Time = 16:12:00, Max vmem = 27.506G
 LSC 1.alpha
LSC 1.alpha