
Single-Molecule Projects
Chaperonins and Protein Aggregation Processes in Amyloid Disease
Protein Aggregation Studies:
Delayed Emergence of Subdiffraction-Sized Mutant Huntingtin Fibrils Following Inclusion Body Formation in Cells
 The identities of toxic aggregate species in Huntington's disease pathogenesis remain ambiguous. While polyQ-expanded huntingtin (Htt) is known to accumulate in compact inclusion bodies inside neurons, this is widely thought to be a protective coping response that sequesters misfolded conformations or aggregated states of the mutated protein. To define the spatial distributions of fluorescently-labeled Htt-exon1 species in the cell model PC12m, we employed highly sensitive single-molecule super-resolution fluorescence imaging. In addition to inclusion bodies and the diffuse pool of monomers and oligomers, small fibrillar aggregates ~100 nm in diameter and up to ~1-2 µm in length were observed for pathogenic polyQ tracts (46 and 97 repeats) after targeted photo-bleaching of the inclusion bodies. These short structures bear a striking resemblance to fibers described in vitro. Definition of the diverse Htt structures in cells will provide an avenue to link the impact of therapeutic agents to aggregate populations and morphologies. The addition of the apical domain of the CCT1 subunit of the eukaryotic chaperonin TRiC/CCT changed the observed distribution of these small fibrillar aggregates.
The identities of toxic aggregate species in Huntington's disease pathogenesis remain ambiguous. While polyQ-expanded huntingtin (Htt) is known to accumulate in compact inclusion bodies inside neurons, this is widely thought to be a protective coping response that sequesters misfolded conformations or aggregated states of the mutated protein. To define the spatial distributions of fluorescently-labeled Htt-exon1 species in the cell model PC12m, we employed highly sensitive single-molecule super-resolution fluorescence imaging. In addition to inclusion bodies and the diffuse pool of monomers and oligomers, small fibrillar aggregates ~100 nm in diameter and up to ~1-2 µm in length were observed for pathogenic polyQ tracts (46 and 97 repeats) after targeted photo-bleaching of the inclusion bodies. These short structures bear a striking resemblance to fibers described in vitro. Definition of the diverse Htt structures in cells will provide an avenue to link the impact of therapeutic agents to aggregate populations and morphologies. The addition of the apical domain of the CCT1 subunit of the eukaryotic chaperonin TRiC/CCT changed the observed distribution of these small fibrillar aggregates.
Steffen J. Sahl, Lana Lau, Willianne I. M. Vonk, Lucien E. Weiss, Judith Frydman and W. E. Moerner, “Delayed emergence of subdiffraction-sized mutant huntingtin fibrils following inclusion body formation in cells,” Quarterly Reviews of Biophysics Discovery (QRB Discovery), pp. 1-13 (2015) doi:10.1017/S0033583515000219. [DOI]
Cellular Inclusion Bodies of Mutant Huntingtin Exon1 Obscure Small Fibrillar Aggregate Species
 The identities of toxic aggregate species in Huntington's disease pathogenesis remain ambiguous. While polyQ-expanded huntingtin (Htt) is known to accumulate in compact inclusion bodies inside neurons, this is widely thought to be a protective coping response that sequesters misfolded conformations or aggregated states of the mutated protein. To define the spatial distributions of fluorescently-labeled Htt-exon1 species in the cell model PC12m, we employed highly sensitive single-molecule super-resolution fluorescence imaging. In addition to inclusion bodies and the diffuse pool of monomers and oligomers, fibrillar aggregates ~100 nm in diameter and up to ~1-2 µm in length were observed for pathogenic polyQ tracts (46 and 97 repeats) after targeted photo-bleaching of the inclusion bodies. These short structures bear a striking resemblance to fibers described in vitro. Definition of the diverse Htt structures in cells will provide an avenue to link the impact of therapeutic agents to aggregate populations and morphologies.
The identities of toxic aggregate species in Huntington's disease pathogenesis remain ambiguous. While polyQ-expanded huntingtin (Htt) is known to accumulate in compact inclusion bodies inside neurons, this is widely thought to be a protective coping response that sequesters misfolded conformations or aggregated states of the mutated protein. To define the spatial distributions of fluorescently-labeled Htt-exon1 species in the cell model PC12m, we employed highly sensitive single-molecule super-resolution fluorescence imaging. In addition to inclusion bodies and the diffuse pool of monomers and oligomers, fibrillar aggregates ~100 nm in diameter and up to ~1-2 µm in length were observed for pathogenic polyQ tracts (46 and 97 repeats) after targeted photo-bleaching of the inclusion bodies. These short structures bear a striking resemblance to fibers described in vitro. Definition of the diverse Htt structures in cells will provide an avenue to link the impact of therapeutic agents to aggregate populations and morphologies.
Steffen J. Sahl, Lucien E. Weiss, Whitney C. Duim, Judith Frydman, and W. E. Moerner, “Cellular Inclusion Bodies of Mutant Huntingtin Exon1 Obscure Small Fibrillar Aggregate Species,” Scientific Reports 2, 895 (2012). [DOI] [![]() Slide]
Slide]
Sub-Diffraction Imaging of Huntingtin Protein Aggregates by Fluorescence Blink-Microscopy and Atomic Force Microscopy
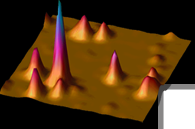
 Huntington's disease arises from an extended polyglutamine region in the huntingtin protein, which leads to huntingtin aggregation in neurons over time. Understanding the morphology of these aggregates with optical microscopy would be useful, but the diffraction limit has prevented such studies until now. Super-resolution optical imaging can be achieved by single-molecule active control microscopy, where in this experiment the control of the emission was provided by forcing the single emitters to blink on and off. Single-molecule super-resolution imaging of fluorescent Huntington’s disease proteins revealed the detailed morphology of aggregates in aqueous environments (color) and showed excellent agreement with atomic force microscopy images (black) of the same aggregates.
Huntington's disease arises from an extended polyglutamine region in the huntingtin protein, which leads to huntingtin aggregation in neurons over time. Understanding the morphology of these aggregates with optical microscopy would be useful, but the diffraction limit has prevented such studies until now. Super-resolution optical imaging can be achieved by single-molecule active control microscopy, where in this experiment the control of the emission was provided by forcing the single emitters to blink on and off. Single-molecule super-resolution imaging of fluorescent Huntington’s disease proteins revealed the detailed morphology of aggregates in aqueous environments (color) and showed excellent agreement with atomic force microscopy images (black) of the same aggregates.
Whitney C. Duim, Bryan Chen, Judith Frydman, and W. E. Moerner, ChemPhysChem, published online 6 July 2011. [![]() Slide] [Journal Link]
Slide] [Journal Link]
Chaperonin Studies:
Sensing Cooperativity in ATP Hydrolysis for Single TRiC/CCT Enzymes in Solution
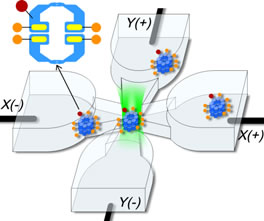 In order to operate in a synchronized fashion, multi-subunit enzymes use cooperative interactions intrinsic to their enzymatic cycle, but this process remains poorly understood. Accordingly, ATP number distributions in various hydrolyzed states have been obtained for single copies of the mammalian double-ring multi-subunit chaperonin TRiC/CCT in free solution using the emission from chaperonin-bound fluorescent nucleotides and closed-loop feedback trapping provided by an Anti-Brownian ELectrokinetic trap (ABEL trap). Observations of the 16-subunit complexes as ADP molecules are dissociating shows a peak in the bound ADP number distribution at 8 ADP, whose height falls over time with little shift in the position of the peak, indicating a highly cooperative ADP release process which would be difficult to observe by ensemble-averaged methods. When AlFx is added to produce ATP hydrolysis transition state mimics (ADP∙AlFx) locked to the complex, the peak at 8 nucleotides dominates for all but the lowest incubation concentrations. Although ensemble averages of the single-molecule data show agreement with standard cooperativity models, surprisingly, the observed number distributions depart from standard models, illustrating the value of these single-molecule observations in constraining the mechanism of cooperativity. While a complete alternative microscopic model cannot be defined at present, the addition of subunit-occupancy-dependent cooperativity in hydrolysis yields distributions consistent with the data.
In order to operate in a synchronized fashion, multi-subunit enzymes use cooperative interactions intrinsic to their enzymatic cycle, but this process remains poorly understood. Accordingly, ATP number distributions in various hydrolyzed states have been obtained for single copies of the mammalian double-ring multi-subunit chaperonin TRiC/CCT in free solution using the emission from chaperonin-bound fluorescent nucleotides and closed-loop feedback trapping provided by an Anti-Brownian ELectrokinetic trap (ABEL trap). Observations of the 16-subunit complexes as ADP molecules are dissociating shows a peak in the bound ADP number distribution at 8 ADP, whose height falls over time with little shift in the position of the peak, indicating a highly cooperative ADP release process which would be difficult to observe by ensemble-averaged methods. When AlFx is added to produce ATP hydrolysis transition state mimics (ADP∙AlFx) locked to the complex, the peak at 8 nucleotides dominates for all but the lowest incubation concentrations. Although ensemble averages of the single-molecule data show agreement with standard cooperativity models, surprisingly, the observed number distributions depart from standard models, illustrating the value of these single-molecule observations in constraining the mechanism of cooperativity. While a complete alternative microscopic model cannot be defined at present, the addition of subunit-occupancy-dependent cooperativity in hydrolysis yields distributions consistent with the data.
Yan Jiang, Nicholai R. Douglas, Nicholas R. Conley, Erik J. Miller, Judith Frydman, and W. E. Moerner, “Sensing Cooperativity in ATP Hydrolysis for Single Multi-Subunit Enzymes in Solution,” Proc. Nat. Acad. Sci. (USA) 108, 16962-16967 (2011), published online 6 September 2011. [![]() Slide] [Journal Link][Commentary]
Slide] [Journal Link][Commentary]
Action of the chaperonin GroEL/ES on a non-native substrate observed with single-molecule FRET
 The double-ring-shaped chaperonin GroEL binds a wide range of non-native polypeptides within its central cavity and, together with its cofactor GroES, assists their folding in an ATP-dependent manner. While the conformational cycle of GroEL/ES has been extensively studied, little is known about how the environment in the central cavity affects substrate conformation. Here we use the von Hippel-Lindau tumor suppressor protein (VHL) as a model substrate for studying the action of the GroEL/ES system on a bound polypeptide. Fluorescent labeling of pairs of sites on VHL for FRET allows VHL to be used to explore how GroEL binding and GroEL/ES/nucleotide binding affect substrate conformation. On average, upon binding to GroEL, all pairs of labeling sites experience compaction relative to the unfolded protein while single-molecule FRET distributions show significant heterogeneity. Upon addition of GroES and ATP to close the GroEL cavity, on average further FRET increases occur between the two hydrophobic regions of VHL, accompanied by FRET decreases between the N- and C-termini. This suggests that ATP- and GroES-induced confinement within the GroEL cavity remodels bound polypeptides by causing expansion (or racking) of some regions and compaction of others, most notably, the hydrophobic core. However, single-molecule observations of the specific FRET changes for individual proteins at the moment of ATP/GroES addition show that a large fraction of the population shows the opposite behavior, that is, FRET decreases between the hydrophobic regions and FRET increases for the N- and C-termini. Our time-resolved single-molecule analysis reveals the underlying heterogeneity of the action of GroES/EL on a bound polypeptide substrate which may arise from the random nature of the specific binding to the various identical subunits of GroEL, and may help explain why multiple rounds of binding and hydrolysis are required for some chaperonin substrates.
The double-ring-shaped chaperonin GroEL binds a wide range of non-native polypeptides within its central cavity and, together with its cofactor GroES, assists their folding in an ATP-dependent manner. While the conformational cycle of GroEL/ES has been extensively studied, little is known about how the environment in the central cavity affects substrate conformation. Here we use the von Hippel-Lindau tumor suppressor protein (VHL) as a model substrate for studying the action of the GroEL/ES system on a bound polypeptide. Fluorescent labeling of pairs of sites on VHL for FRET allows VHL to be used to explore how GroEL binding and GroEL/ES/nucleotide binding affect substrate conformation. On average, upon binding to GroEL, all pairs of labeling sites experience compaction relative to the unfolded protein while single-molecule FRET distributions show significant heterogeneity. Upon addition of GroES and ATP to close the GroEL cavity, on average further FRET increases occur between the two hydrophobic regions of VHL, accompanied by FRET decreases between the N- and C-termini. This suggests that ATP- and GroES-induced confinement within the GroEL cavity remodels bound polypeptides by causing expansion (or racking) of some regions and compaction of others, most notably, the hydrophobic core. However, single-molecule observations of the specific FRET changes for individual proteins at the moment of ATP/GroES addition show that a large fraction of the population shows the opposite behavior, that is, FRET decreases between the hydrophobic regions and FRET increases for the N- and C-termini. Our time-resolved single-molecule analysis reveals the underlying heterogeneity of the action of GroES/EL on a bound polypeptide substrate which may arise from the random nature of the specific binding to the various identical subunits of GroEL, and may help explain why multiple rounds of binding and hydrolysis are required for some chaperonin substrates.
S. Y. Kim, E. J. Miller, J. Frydman, and W. E. Moerner, J. Molec. Biol. 401, 553-563 (2010), published online 30 June 2010. [![]() Slide] [Journal Link]
Slide] [Journal Link]
Probing Nucleotide-Induced Cavity Polarity Changes in Molecular Chaperones
 Hydrophobic interactions play a major role in binding non-native substrate proteins in the central cavity of the bacterial chaperonin GroEL. The sequence of local conformational changes by which GroEL and its cofactor GroES assist protein folding can be explored using the polarity-sensitive fluorescence probe Nile Red. A specific single-cysteine mutant of GroEL (Cys261), whose cysteine is located inside the central cavity at the apical region of the protein, was covalently labeled with synthetically prepared Nile Red maleimide (NR). Bulk fluorescence spectra of Cys261-NR were measured to examine the effects of binding of the stringent substrate, malate dehydrogenase (MDH), GroES, and nucleotide on the local environment of the probe. After binding denatured substrate, the fluorescence intensity increased by 32±7%, suggesting enhanced hydrophobicity at the position of the label. On the other hand, in the presence of ATP, the fluorescence intensity decreased by 13±3%, implying increased local polarity. In order to explore the sequence of local polarity changes, substrate, GroES, and various nucleotides were added in different orders; the resulting changes in emission intensity provide insight into the sequence of conformational changes occurring during GroEL-mediated protein folding.
Hydrophobic interactions play a major role in binding non-native substrate proteins in the central cavity of the bacterial chaperonin GroEL. The sequence of local conformational changes by which GroEL and its cofactor GroES assist protein folding can be explored using the polarity-sensitive fluorescence probe Nile Red. A specific single-cysteine mutant of GroEL (Cys261), whose cysteine is located inside the central cavity at the apical region of the protein, was covalently labeled with synthetically prepared Nile Red maleimide (NR). Bulk fluorescence spectra of Cys261-NR were measured to examine the effects of binding of the stringent substrate, malate dehydrogenase (MDH), GroES, and nucleotide on the local environment of the probe. After binding denatured substrate, the fluorescence intensity increased by 32±7%, suggesting enhanced hydrophobicity at the position of the label. On the other hand, in the presence of ATP, the fluorescence intensity decreased by 13±3%, implying increased local polarity. In order to explore the sequence of local polarity changes, substrate, GroES, and various nucleotides were added in different orders; the resulting changes in emission intensity provide insight into the sequence of conformational changes occurring during GroEL-mediated protein folding.
So Yeon Kim, Alexander N. Semyonov, Robert J. Twieg, Arthur L. Horwich, Judith Frydman, and W. E. Moerner, "Probing the Sequence of Conformationally-Induced Polarity Changes in the Molecular Chaperonin GroEL with Fluorescence Spectroscopy," J. Phys. Chem. B 109, 24517-24525 (2005). [![]() Full Text] [Slide]
Full Text] [Slide]
![]() The Moerner Laboratory has been a member of the Center for Protein Folding Machinery, and has collaborated over the years with the groups of Judith Frydman at Stanford, Wah Chiu at Baylor College of Medicine, Bill Mobley at UCSD, and Leslie Thompson at UCI.
The Moerner Laboratory has been a member of the Center for Protein Folding Machinery, and has collaborated over the years with the groups of Judith Frydman at Stanford, Wah Chiu at Baylor College of Medicine, Bill Mobley at UCSD, and Leslie Thompson at UCI.