
Single-Molecule Projects:
Applications of Superresolution Imaging in 2D and 3D Using Single-Molecule Active-Control Microscopy (SMACM), STED, and Correlative cryo-EM in Bacterial and Mammalian Cells
Current Subgroup Members: Dr. Leonid Andronov, Dr. Pierre Jouchet , Ashwin Balaji, Davis Perez, Dr. Peter Dahlberg (SLAC)
RECENT ADVANCES - scroll down!
Basics
Optical fluorescence microscopy is an important tool for cell biology because light can be used to non-invasively probe a sample with relatively small perturbation of the specimen, enabling dynamical observation of the motions of internal structures in living cells, but with resolution usually limited to ~250 nm by optical diffraction. Single-molecule epifluorescence microscopy achieves nanometer-scale resolution by taking advantage of the fact that the point spread function of an isolated nanoscale emitter can be fit to a precision far greater than the standard diffraction limit. Over the past few years, the utility of this technique has been extended to the regime of biologically relevant, room-temperature experiments with clever use of photoactivation or other active control schemes to control the emitting concentration of single nanoscale fluorescent labels (PALM, F-PALM, STORM).1
All such superresolution techniques are based on the critical requirement of imaging nanometer-sized single-molecule emitters and on the use of an active control mechanism to produce sparse sub-ensembles. For convenience, we refer to these techniques as a group using the inclusive, general term, Single-Molecule Active-Control Microscopy (SMACM). In SMACM experiments, structures labeled by an ensemble of photoactivatable fluorophores too dense to be imaged simultaneously are resolved over repeated cycles in each of which only a sparse subset of the fluorophores is activated. The final superresolution image is reconstituted from a superposition of single-molecule (low-concentration) images.
An alternative method for obtaining superresolution is to use STED (Stimulated Emission Depletion Microscopy) invented by Stephan Hell and its variants2, where a donut-shaped beam with a dark hole in the middle is used to quench or turn off the emission of molecules outside the dark region. Implemented in a confocal scanning microscope, this method also produces resolution beyond the optical diffraction limit. Using our home-built STED microscope, we have explored centriolar protein structures and chromatin organization in various cellular systems beyond the diffraction limit..
We have pursued several thrusts in collaboration with our colleagues:
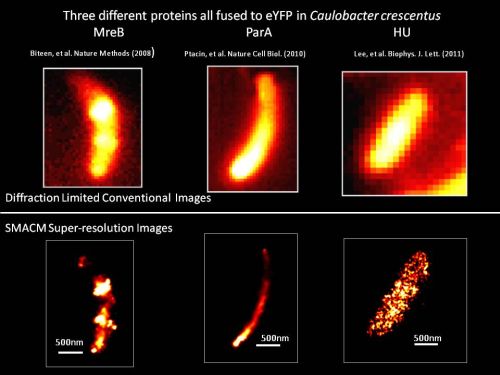
- Direct superresolution imaging of protein localization patterns in bacterial cells in collaboration with the group of Prof. Lucy Shapiro, Stanford, with emphasis on polar protein superstructure and organization.
- Imaging of nanoscale protein superstructures in 3D in cells.
- Single-molecule tracking and imaging as a probe of cellular signaling in the primary cilium.
- Nuclear chromatin organization.
- Imaging of the glycocalyx.
- Correlative super-resolution microscopy and electron microscopy.
- Structures of viral RNA and viral proteins in SARS-CoV-2 infected cells
EXAMPLE:
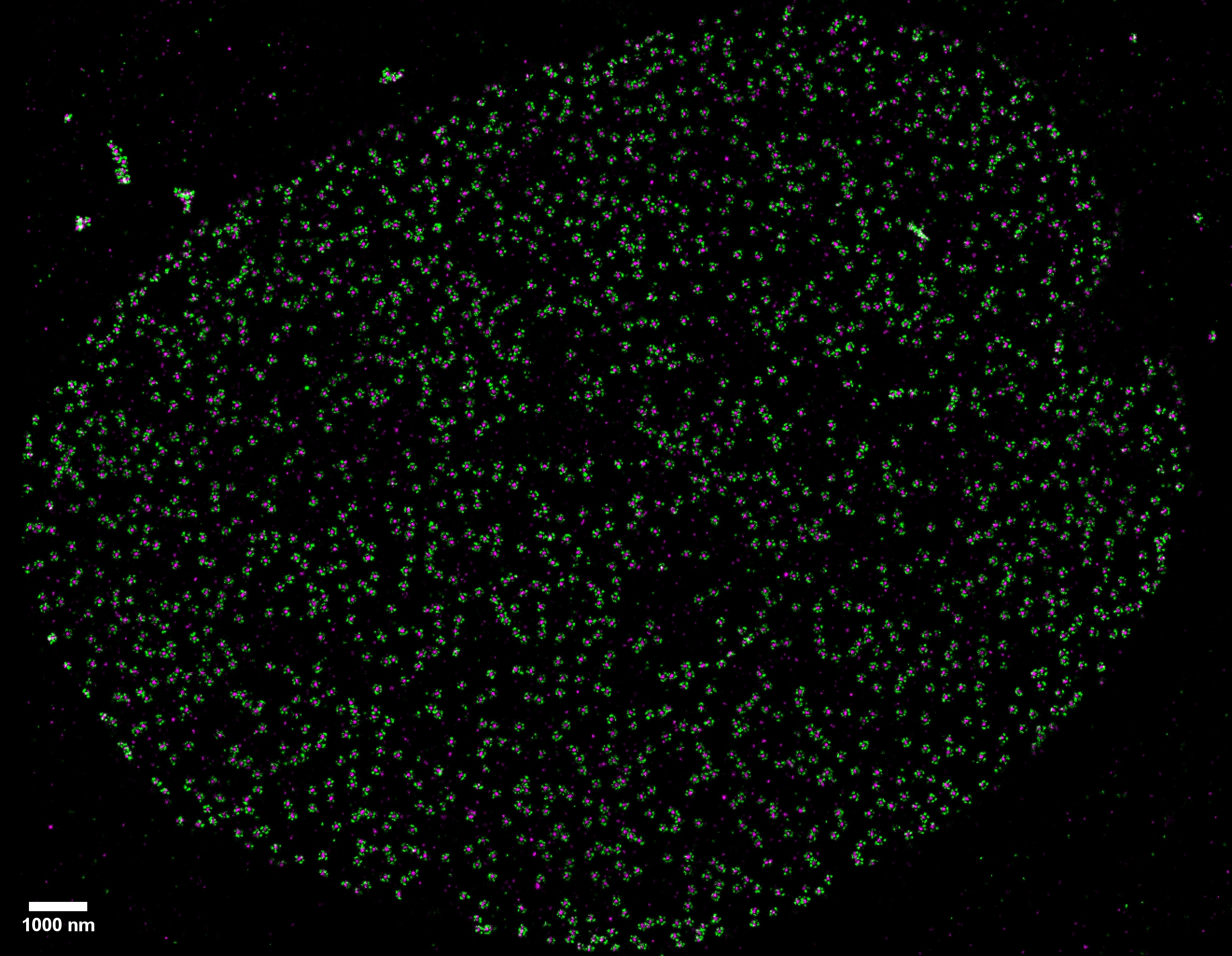 Two-color (d)STORM super-resolution image of nuclear pores on the nuclear envelope in a HeLa cell. The nuclear pore complex protein Nup133 (green) was detected with CF583R-conjugated secondary antibodies (DOL = 4.18), and Nup62 (magenta) was detected with Alexa Fluor 647-conjugated secondary antibodies. (d)STORM was performed in an oxygen scavenging imaging buffer containing 50 mM MEA. Courtesy of Dr. Leonid Andronov, Moerner Laboratory, Stanford University.
Two-color (d)STORM super-resolution image of nuclear pores on the nuclear envelope in a HeLa cell. The nuclear pore complex protein Nup133 (green) was detected with CF583R-conjugated secondary antibodies (DOL = 4.18), and Nup62 (magenta) was detected with Alexa Fluor 647-conjugated secondary antibodies. (d)STORM was performed in an oxygen scavenging imaging buffer containing 50 mM MEA. Courtesy of Dr. Leonid Andronov, Moerner Laboratory, Stanford University.
Recent Reviews:
Peter D. Dahlberg and W. E. Moerner, “Cryogenic Super-Resolution Fluorescence and Electron Microscopy Correlated at the Nanoscale,” Ann. Revs. Phys. Chem. 72 (April 2021) (DOI: 10.1146/annurev-physchem-090319-051546, published online 13 January 2021).
W. E. Moerner, "Viewpoint: Single Molecules at 31: What's Next?" Nano Lett 20, 8427-8429 (2020) (DOI: 10.1021/acs.nanolett.0c04042, published online 10 November 2020).
Leonhard Möckl and W. E. Moerner, Super-resolution microscopy with single molecules in biology and beyond essentials, current trends, and future challenges, Perspective Article, J. Am. Chem. Soc. 142, 17828-17844 (2020) (DOI: 10.1021/JACS.0c08178, published online 9 October 2020).
Mikael P. Backlund, Matthew D. Lew, Adam S. Backer, Steffen J. Sahl, and W. E. Moerner, “The role of molecular dipole orientation in single-molecule fluorescence microscopy and implications for super-resolution imaging,” Minireview, ChemPhysChem , published online December 30, 2013. DOI
Andreas Gahlmann and W. E. Moerner, “Exploring bacterial cell biology with single-molecule tracking and super-resolution imaging,” Nature Reviews Microbiology 12, 9-22 (2014), published online December 16, 2013. DOI
Steffen J. Sahl and W. E. Moerner, "Super-resolution Fluorescence Imaging with Single Molecules,” Curr. Opin. Struct. Biol. 23, 778-787 (2013), published online 8 August 2013. DOI
W. E. Moerner, “Microscopy beyond the diffraction limit using actively controlled single molecules,” J. Microsc. 246, 213-220 (2012), published online 12 April 2012. DOI
Matthew D. Lew, Steven F. Lee, Michael A. Thompson, Hsiao-lu D. Lee, and W. E. Moerner, “Single-Molecule Photocontrol and Nanoscopy,” in Far-Field Optical Nanoscopy, P. Tinnefeld, C. Eggeling, and S. W. Hell, Eds., Springer Series on Fluorescence (Springer, Berlin, Heidelberg, 2012), published online 21 February 2012. DOI
J. S. Biteen, L. Shapiro, and W. E. Moerner, “Exploring Protein Superstructures and Dynamics in Live Bacterial Cells Using Single-Molecule and Superresolution Imaging,” Ch. 8 of Single-Molecule Techniques: Methods and Protocols, E. J. G. Peterman and G. J. L. Wuite, Eds., Methods in Molecular Biology Volume 783 (Humana Press, New York, 2011), pp. 139-158. DOI
M. A. Thompson, J. S. Biteen, S. J. Lord, N. R. Conley, and W. E. Moerner, “Molecules and Methods for Super-Resolution Imaging,” Methods in Enzymology, Volume 475, Nils G. Walter, Editor (Elsevier, New York, 2010), Chapter 2, pp. 27-59. DOI
J. S. Biteen and W. E. Moerner, “Single-Molecule and Superresolution Imaging in Live Bacterial Cells,” in Cell Biology of Bacteria, L. Shapiro and R. Losick, Eds., Cold Spring Harbor Perspectives in Biology 2010; 2:a000448 (Cold Spring Harbor Laboratory Press, 2011), first published online February 3, 2010.
Recent Advances:
Nanoscale cellular organization of viral RNA and proteins in SARS-CoV-2 replication organelles
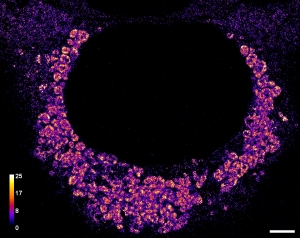 The SARS-CoV-2 viral infection transforms host cells and produces special organelles in many ways, and we focus on the replication organelles, the sites of replication of viral genomic RNA (vgRNA). To date, the precise cellular localization of key RNA molecules and replication intermediates has been elusive in electron microscopy studies. We use super-resolution fluorescence microscopy and specific labeling to reveal the nanoscopic organization of replication organelles that contain numerous vgRNA molecules along with the replication enzymes and clusters of viral double-stranded RNA (dsRNA). We show that the replication organelles are organized differently at early and late stages of infection. Surprisingly, vgRNA accumulates into distinct globular clusters in the cytoplasmic perinuclear region, which grow and accommodate more vgRNA molecules as infection time increases. The localization of endoplasmic reticulum (ER) markers and nsp3 (a component of the double-membrane vesicle, DMV) at the periphery of the vgRNA clusters suggests that replication organelles are encapsulated into DMVs, which have membranes derived from the host ER. These organelles merge into larger vesicle packets as infection advances. Precise co-imaging of the nanoscale cellular organization of vgRNA, dsRNA, and viral proteins in replication organelles of SARS-CoV-2 may inform therapeutic approaches that target viral replication and associated processes.
The SARS-CoV-2 viral infection transforms host cells and produces special organelles in many ways, and we focus on the replication organelles, the sites of replication of viral genomic RNA (vgRNA). To date, the precise cellular localization of key RNA molecules and replication intermediates has been elusive in electron microscopy studies. We use super-resolution fluorescence microscopy and specific labeling to reveal the nanoscopic organization of replication organelles that contain numerous vgRNA molecules along with the replication enzymes and clusters of viral double-stranded RNA (dsRNA). We show that the replication organelles are organized differently at early and late stages of infection. Surprisingly, vgRNA accumulates into distinct globular clusters in the cytoplasmic perinuclear region, which grow and accommodate more vgRNA molecules as infection time increases. The localization of endoplasmic reticulum (ER) markers and nsp3 (a component of the double-membrane vesicle, DMV) at the periphery of the vgRNA clusters suggests that replication organelles are encapsulated into DMVs, which have membranes derived from the host ER. These organelles merge into larger vesicle packets as infection advances. Precise co-imaging of the nanoscale cellular organization of vgRNA, dsRNA, and viral proteins in replication organelles of SARS-CoV-2 may inform therapeutic approaches that target viral replication and associated processes.
Leonid Andronov*, Mengting Han*, Yanyu Zhu, Anish R. Roy, Andrew E. S. Barentine, Jaishree Garhyan, Lei S. Qi, and W.E. Moerner, (*equal contributions) Nanoscale cellular organization of viral RNA and proteins in SARS-CoV-2 replication organelles, Nature Commun. (2024) 15: 4644 (DOI: /10.1038/s41467-024-48991-x, published online 31 May 2024). Stanford Report News Article
Exploring Transient States of PAmKate to Enable Improved Cryogenic Single-Molecule Imaging
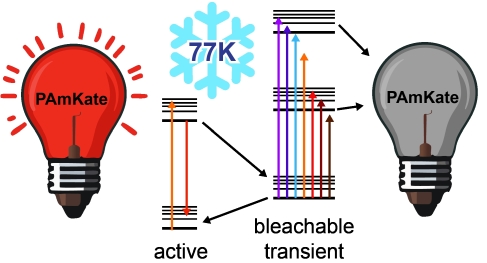 Super-resolved cryogenic correlative light and electron microscopy is a powerful approach which combines the single-molecule specificity and sensitivity of fluorescence imaging with the nano-scale resolution of cryogenic electron tomography. Key to this method is active control over the emissive state of fluorescent labels to ensure sufficient sparsity to localize individual emitters. Recent work has identified fluorescent proteins (FPs) which photoactivate or photoswitch efficiently at cryogenic temperatures, but long on-times due to reduced quantum yield of photobleaching remains a challenge for imaging structures with a high density of localizations. In this work, we explore the photophysical properties of the red photoactivatable FP PAmKate and identify a 2-color process leading to enhanced turn-off of active emitters, improving localization rate. Specifically, after excitation of ground state molecules, we find a transient state forms with a lifetime of ~2 ms under cryogenic conditions which can be bleached by exposure to a second wavelength. We measure the response of the transient state to different wavelengths, demonstrate how this mechanism can be used to improve imaging, and provide a blueprint for study of other FPs at cryogenic temperatures.
Super-resolved cryogenic correlative light and electron microscopy is a powerful approach which combines the single-molecule specificity and sensitivity of fluorescence imaging with the nano-scale resolution of cryogenic electron tomography. Key to this method is active control over the emissive state of fluorescent labels to ensure sufficient sparsity to localize individual emitters. Recent work has identified fluorescent proteins (FPs) which photoactivate or photoswitch efficiently at cryogenic temperatures, but long on-times due to reduced quantum yield of photobleaching remains a challenge for imaging structures with a high density of localizations. In this work, we explore the photophysical properties of the red photoactivatable FP PAmKate and identify a 2-color process leading to enhanced turn-off of active emitters, improving localization rate. Specifically, after excitation of ground state molecules, we find a transient state forms with a lifetime of ~2 ms under cryogenic conditions which can be bleached by exposure to a second wavelength. We measure the response of the transient state to different wavelengths, demonstrate how this mechanism can be used to improve imaging, and provide a blueprint for study of other FPs at cryogenic temperatures.
Davis Perez, Dara P. Dowlatshahi, Christopher A. Azaldegui, T. Bertie Ansell, Peter D. Dahlberg, and W. E. Moerner, Exploring Transient States of PAmKate to Enable Improved Cryogenic Single-Molecule Imaging, Journal of the American Chemical Society (citation tbd) (DOI: 10.1021/jacs.4c05632, published online 10 October 2024).
Characterization of mApple as a Red Fluorescent Protein for Cryogenic Single-Molecule Imaging with Turn-off and Turn-on Active Control Mechanisms
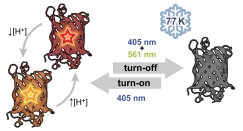 Single-molecule superresolution microscopy is a powerful tool for the study of biological structures on size scales smaller than the optical diffraction limit. Imaging samples at cryogenic temperatures (77 K) reduces the quantum yield of photobleaching for many fluorescent labels, yielding localization precisions below 10 nm. Cryogenic imaging further enables correlation with cryogenic electron tomography. A key limitation in applying methods such as PALM and STORM to samples maintained at 77 K is the limited number of fluorophores known to undergo efficient turn-on and turn-off mechanisms necessary to control the sparsity of active emitters. We find that mApple, a red-emitting fluorescent protein, undergoes a novel turn-off mechanism in response to simultaneous illumination with two colors of light. This turn-off mechanism enables localization of many individual molecules in initially bright samples, but the final density of localizable emitters is limited by relatively inefficient turn-on (photoactivation). Bulk excitation and emission spectroscopy shows that mApple has access to two distinct emissive states as well as dark states accessible optically or through changes in pH. The bright and stable emission of mApple enables widefield collection of single-molecule emission spectra, which highlight the complex nature and environmental sensitivity of states observed in red fluorescent proteins.
Single-molecule superresolution microscopy is a powerful tool for the study of biological structures on size scales smaller than the optical diffraction limit. Imaging samples at cryogenic temperatures (77 K) reduces the quantum yield of photobleaching for many fluorescent labels, yielding localization precisions below 10 nm. Cryogenic imaging further enables correlation with cryogenic electron tomography. A key limitation in applying methods such as PALM and STORM to samples maintained at 77 K is the limited number of fluorophores known to undergo efficient turn-on and turn-off mechanisms necessary to control the sparsity of active emitters. We find that mApple, a red-emitting fluorescent protein, undergoes a novel turn-off mechanism in response to simultaneous illumination with two colors of light. This turn-off mechanism enables localization of many individual molecules in initially bright samples, but the final density of localizable emitters is limited by relatively inefficient turn-on (photoactivation). Bulk excitation and emission spectroscopy shows that mApple has access to two distinct emissive states as well as dark states accessible optically or through changes in pH. The bright and stable emission of mApple enables widefield collection of single-molecule emission spectra, which highlight the complex nature and environmental sensitivity of states observed in red fluorescent proteins.
Annina M. Sartor, Peter D. Dahlberg, Davis Perez, and W. E. Moerner,"Characterization of mApple as a Red Fluorescent Protein for Cryogenic Single-Molecule Imaging with Turn-Off and Turn-On Active Control Mechanisms," Part of Special Issue Early-Career and Emerging Researchers in Physical Chemistry Volume 2, J. Phys. Chem B 172, 2690-2700 (2023) (DOI:10.1021/acs.jpcb.2c08995, published online 21 March 2023).
Nanometer-scale distribution of PD-1 in the melanoma tumor microenvironment
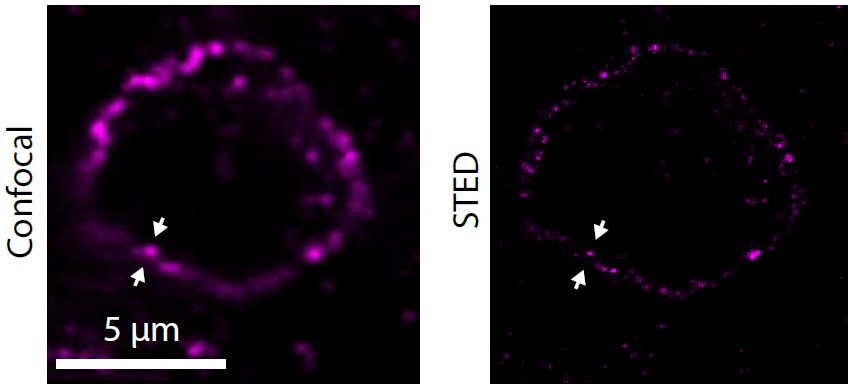 The nanometer-scale spatial organization of immune receptors plays a role in cell activation and suppression. While the connection between this spatial organization and cell signaling events is emerging from cell culture experiments, how these results translate to more physiologically relevant settings like the tumor microenvironment remains poorly understood due to the challenges of high-resolution imaging in vivo. Here we perform super-resolution immunofluorescence microscopy of human melanoma tissue sections to examine the spatial organization of the immune checkpoint inhibitor programmed cell death 1 (PD-1). We show that PD-1 exhibits a variety of organizations ranging from nanometer-scale clusters to more uniform membrane labeling. Our results demonstrate the capability of super-resolution imaging to examine the spatial organization of immune checkpoint markers in the tumor microenvironment, suggesting future direction for both clinical and immunology research.
The nanometer-scale spatial organization of immune receptors plays a role in cell activation and suppression. While the connection between this spatial organization and cell signaling events is emerging from cell culture experiments, how these results translate to more physiologically relevant settings like the tumor microenvironment remains poorly understood due to the challenges of high-resolution imaging in vivo. Here we perform super-resolution immunofluorescence microscopy of human melanoma tissue sections to examine the spatial organization of the immune checkpoint inhibitor programmed cell death 1 (PD-1). We show that PD-1 exhibits a variety of organizations ranging from nanometer-scale clusters to more uniform membrane labeling. Our results demonstrate the capability of super-resolution imaging to examine the spatial organization of immune checkpoint markers in the tumor microenvironment, suggesting future direction for both clinical and immunology research.
Colin J. Comerci, Dannielle G. McCarthy, Mehdi Nosrati, Kevin B. Kim, Mohammed Kashani-Sabet, W. E. Moerner, and Stanley P. Leong, Nanometer-scale distribution of PD-1 in the melanoma tumor microenvironment, J. Radiol. Oncol. 7, 020-025 (2023) (DOI: 10.29328/journal.jro.1001048, published online 10 May 2023).
Structural and photophysical characterization of the small ultra-red fluorescent protein smURFP
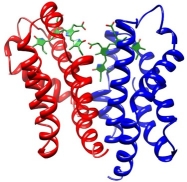 The small Ultra-Red Fluorescent Protein (smURFP) represents a new class of fluorescent protein with exceptional photostability and brightness derived from allophycocyanin in a previous directed evolution. Here, we report the smURFP crystal structure to better understand properties and enable further engineering of improved variants. We compare this structure to the structures of allophycocyanin and smURFP mutants to identify the structural origins of the molecular brightness. We then use a structure-guided approach to develop monomeric smURFP variants that fluoresce with phycocyanobilin but not biliverdin. Furthermore, we measure smURFP photophysical properties necessary for advanced imaging modalities, such as those relevant for two-photon, fluorescence lifetime, and single-molecule imaging. We observe that smURFP has the largest two-photon cross-section measured for a fluorescent protein, and that it produces more photons than organic dyes. Altogether, this study expands our understanding of the smURFP, which will inform future engineering toward optimal FPs compatible with whole organism studies.
The small Ultra-Red Fluorescent Protein (smURFP) represents a new class of fluorescent protein with exceptional photostability and brightness derived from allophycocyanin in a previous directed evolution. Here, we report the smURFP crystal structure to better understand properties and enable further engineering of improved variants. We compare this structure to the structures of allophycocyanin and smURFP mutants to identify the structural origins of the molecular brightness. We then use a structure-guided approach to develop monomeric smURFP variants that fluoresce with phycocyanobilin but not biliverdin. Furthermore, we measure smURFP photophysical properties necessary for advanced imaging modalities, such as those relevant for two-photon, fluorescence lifetime, and single-molecule imaging. We observe that smURFP has the largest two-photon cross-section measured for a fluorescent protein, and that it produces more photons than organic dyes. Altogether, this study expands our understanding of the smURFP, which will inform future engineering toward optimal FPs compatible with whole organism studies.
Atanu Maiti, Cosmo Z. Buffalo, Saumya Saurabh, Felipe Montecinos-Franjola, Justin S. Hachey, William J. Conlon, Geraldine N. Tran, Mikhail Drobizhev, W. E. Moerner, Partho Ghosh, Hiroshi Matsuo, Roger Y. Tsien, John Y. Lin, and Erik A. Rodriguez, Structural and photophysical characterization of the small ultra-red fluorescent protein, Nature Commun. 14, 4155 (2023), (DOI: 10.1038/s41467-023-39776-9, published online 12 July 2023).
Metallic support films reduce optical heating in cryogenic correlative light and electron tomography
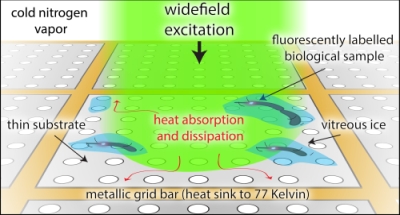 Super-resolved cryogenic correlative light and electron tomography is an emerging method that provides both the single-molecule sensitivity and specificity of fluorescence imaging, and the molecular scale resolution and detailed cellular context of tomography, all in vitrified cells preserved in their native hydrated state. Technical hurdles that limit these correlative experiments need to be overcome for the full potential of this approach to be realized. Chief among these is sample heating due to optical excitation which leads to devitrification, a phase transition from amorphous to crystalline ice. Here we show that much of this heating is due to the material properties of the support film of the electron microscopy grid, specifically the absorptivity and thermal conductivity. We demonstrate through experiment and simulation that the properties of the standard holey carbon electron microscopy grid lead to substantial heating under optical excitation. In order to avoid devitrification, optical excitation intensities must be kept orders of magnitude lower than the intensities commonly employed in room temperature super-resolution experiments. We further show that the use of metallic films, either holey gold grids, or custom made holey silver grids, alleviate much of this heating. For example, the holey silver grids permit 20? the optical intensities used on the standard holey carbon grids. Super-resolution correlative experiments conducted on holey silver grids under these increased optical excitation intensities have a corresponding increase in the rate of single-molecule fluorescence localizations. This results in an increased density of localizations and improved correlative imaging without deleterious effects from sample heating.
Super-resolved cryogenic correlative light and electron tomography is an emerging method that provides both the single-molecule sensitivity and specificity of fluorescence imaging, and the molecular scale resolution and detailed cellular context of tomography, all in vitrified cells preserved in their native hydrated state. Technical hurdles that limit these correlative experiments need to be overcome for the full potential of this approach to be realized. Chief among these is sample heating due to optical excitation which leads to devitrification, a phase transition from amorphous to crystalline ice. Here we show that much of this heating is due to the material properties of the support film of the electron microscopy grid, specifically the absorptivity and thermal conductivity. We demonstrate through experiment and simulation that the properties of the standard holey carbon electron microscopy grid lead to substantial heating under optical excitation. In order to avoid devitrification, optical excitation intensities must be kept orders of magnitude lower than the intensities commonly employed in room temperature super-resolution experiments. We further show that the use of metallic films, either holey gold grids, or custom made holey silver grids, alleviate much of this heating. For example, the holey silver grids permit 20? the optical intensities used on the standard holey carbon grids. Super-resolution correlative experiments conducted on holey silver grids under these increased optical excitation intensities have a corresponding increase in the rate of single-molecule fluorescence localizations. This results in an increased density of localizations and improved correlative imaging without deleterious effects from sample heating.
Peter D. Dahlberg, Davis Perez, Corey W. Hecksel, Wah Chiu, and W.E. Moerner, Metallic support films reduce optical heating in cryogenic correlative light and electron tomography, J. Struct. Biol. 214, 107901 (2022) (DOI: 10.1016/j.jsb.2022.107901, published online 1 October 2022).
Identification and demonstration of roGFP2 as an environmental sensor for cryogenic correlative light and electron microscopy
 Cryogenic correlative light and electron microscopy (cryo-CLEM) seeks to leverage orthogonal information present in two powerful imaging modalities. While recent advances in cryogenic electron microscopy (cryo-EM) allow for the visualization and identification of structures within cells at the nanometer scale, information regarding the cellular environment, such as pH, membrane potential, ionic strength etc. that influence the observed structures remains absent. Fluorescence microscopy can potentially be used to reveal this information when specific labels, known as fluorescent biosensors, are used, but there has been minimal use of such biosensors in cryo-CLEM to date. Here we demonstrate the applicability of one such biosensor, the fluorescent protein roGFP2, for cryo-CLEM experiments. At room temperature, the ratio of roGFP2 emission brightness when excited at 425 nm or 488 nm is known to report on the local redox potential. When samples containing roGFP2 are rapidly cooled to 77K in a manner compatible with cryo-EM, the ratio of excitation peaks remains a faithful indicator of the redox potential at the time of freezing. Using purified protein in different oxidizing/reducing environments, we generate a calibration curve which can be used to analyze in situ measurements. As a proof-of-principle demonstration, we investigate the oxidation/reduction state within vitrified Caulobacter crescentus cells.
Cryogenic correlative light and electron microscopy (cryo-CLEM) seeks to leverage orthogonal information present in two powerful imaging modalities. While recent advances in cryogenic electron microscopy (cryo-EM) allow for the visualization and identification of structures within cells at the nanometer scale, information regarding the cellular environment, such as pH, membrane potential, ionic strength etc. that influence the observed structures remains absent. Fluorescence microscopy can potentially be used to reveal this information when specific labels, known as fluorescent biosensors, are used, but there has been minimal use of such biosensors in cryo-CLEM to date. Here we demonstrate the applicability of one such biosensor, the fluorescent protein roGFP2, for cryo-CLEM experiments. At room temperature, the ratio of roGFP2 emission brightness when excited at 425 nm or 488 nm is known to report on the local redox potential. When samples containing roGFP2 are rapidly cooled to 77K in a manner compatible with cryo-EM, the ratio of excitation peaks remains a faithful indicator of the redox potential at the time of freezing. Using purified protein in different oxidizing/reducing environments, we generate a calibration curve which can be used to analyze in situ measurements. As a proof-of-principle demonstration, we investigate the oxidation/reduction state within vitrified Caulobacter crescentus cells.
Davis Perez, Peter D. Dahlberg, Jiarui Wang, Annina M. Sartor, Julia S. Borden, Lucy Shapiro, and W.E. Moerner, Identification and demonstration of roGFP2 as an environmental sensor for cryogenic correlative light and electron microscopy, J. Struct. Biol. 214, 107881 (2022) (DOI: 10.1016/j.jsb.2022.107881, published online 8 July 2022).
Multi-color super-resolution imaging to study human coronavirus RNA during cellular infection
 MOTIVATION: SARS-CoV-2 is one of the seven coronaviruses known to infect humans, exhibiting wide-spread respiratory syndromes with severe and often fatal disease progression. The subcellular spatial organization of viral components and their interaction with cellular compartments are critical to the infection process. While the scientific community has studied coronavirus biology using genomics, biochemistry, cryo-EM, and electron tomography, how coronavirus RNA is spatially organized in the cell at the different stages of the viral replication cycle at the nanoscale resolution is largely unknown. We develop a multi-color super-resolution microscopy imaging framework to examine the spatial interactions between viral RNA and viral factors during host cell infection. Our imaging framework will support future investigations of future coronavirus biology and therapeutic effects on the 20 nm spatial scale.
MOTIVATION: SARS-CoV-2 is one of the seven coronaviruses known to infect humans, exhibiting wide-spread respiratory syndromes with severe and often fatal disease progression. The subcellular spatial organization of viral components and their interaction with cellular compartments are critical to the infection process. While the scientific community has studied coronavirus biology using genomics, biochemistry, cryo-EM, and electron tomography, how coronavirus RNA is spatially organized in the cell at the different stages of the viral replication cycle at the nanoscale resolution is largely unknown. We develop a multi-color super-resolution microscopy imaging framework to examine the spatial interactions between viral RNA and viral factors during host cell infection. Our imaging framework will support future investigations of future coronavirus biology and therapeutic effects on the 20 nm spatial scale.
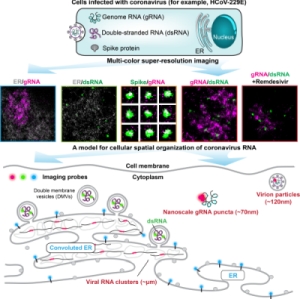
SUMMARY:The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the third human coronavirus within 20 years that gave rise to a life-threatening disease and the first to reach pandemic spread. To make therapeutic headway against current and future coronaviruses, the biology of coronavirus RNA during infection must be precisely understood. Here, we present a robust and generalizable framework combining high-throughput confocal and super-resolution microscopy imaging to study coronavirus infection at the nanoscale. Employing the model human coronavirus HCoV-229E, we specifically labeled coronavirus genomic RNA (gRNA) and double-stranded RNA (dsRNA) via multicolor RNA-immunoFISH and visualized their localization patterns within the cell. The 20nm resolution achieved by of our approach uncovers a striking spatial organization of gRNA and dsRNA into three distinct structures and enables quantitative characterization of the status of the infection after antiviral drug treatment. Our approach provides a comprehensive imaging framework that will enable future investigations of coronavirus fundamental biology and therapeutic effects.
Jiarui Wang, Mengting Han, Anish R. Roy, Haifeng Wang, Leonhard Möckl, Leiping Zeng, W.E. Moerner, and Lei S. Qi, Multi-color super-resolution imaging to study human coronavirus RNA during cellular infection, Cell Reports Methods 2, 100170 (2022) (DOI: 10.1016/j.crmeth.2022.100170, published online 28 February 2022).
ATP-responsive biomolecular condensates tune bacterial kinase signaling-the Story of SpmX, DivJ, and PopZ
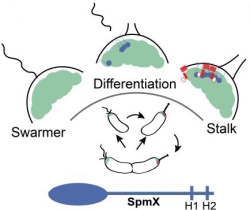 Biomolecular condensates formed via liquid-liquid phase separation enable spatial and temporal organization of enzyme activity. Phase separation in many eukaryotic condensates has been shown to be responsive to intracellular Adenosine triphosphate (ATP) levels, although the consequences of such mechanisms for enzymes sequestered within the condensates are unknown. Here, we show that ATP depletion promotes phase separation in bacterial condensates composed of intrinsically disordered proteins. Enhanced phase separation promotes the sequestration and activity of a client kinase enabling robust signaling and maintenance of viability under the stress posed by nutrient scarcity. We propose that a diverse repertoire of condensates can serve as control knobs to tune enzyme sequestration and reactivity in response to the metabolic state of bacterial cells.
Biomolecular condensates formed via liquid-liquid phase separation enable spatial and temporal organization of enzyme activity. Phase separation in many eukaryotic condensates has been shown to be responsive to intracellular Adenosine triphosphate (ATP) levels, although the consequences of such mechanisms for enzymes sequestered within the condensates are unknown. Here, we show that ATP depletion promotes phase separation in bacterial condensates composed of intrinsically disordered proteins. Enhanced phase separation promotes the sequestration and activity of a client kinase enabling robust signaling and maintenance of viability under the stress posed by nutrient scarcity. We propose that a diverse repertoire of condensates can serve as control knobs to tune enzyme sequestration and reactivity in response to the metabolic state of bacterial cells.
Saumya Saurabh, Trisha N. Chong, Camille Bayas, Peter D. Dahlberg, Heather N. Cartwright, W. E. Moerner, and Lucy Shapiro, ATP-responsive biomolecular condensates tune bacterial kinase signaling, Sci. Advances 8, eabm6570 (2022) (DOI: 10.1126/sciadv.abm6570, published online 16 February 2022).
A localized adaptor protein, PopA, performs distinct functions at the Caulobacter cell poles
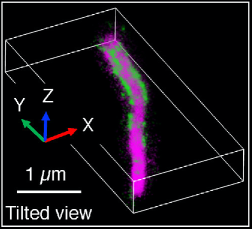
 Asymmetric cell division generates two daughter cells with distinct characteristics and fates. Positioning different regulatory and signaling proteins at the opposing ends of the predivisional cell produces molecularly distinct daughter cells. Here, we report a new strategy deployed by the asymmetrically dividing bacterium Caulobacter crescentus where a regulatory protein is programed to perform distinct functions at the opposing cell poles. We find that the CtrA proteolysis adaptor protein PopA assumes distinct oligomeric states at the two cell poles through asymmetrically distributed c-di-GMP: dimeric at the stalked pole and monomeric at the swarmer pole. Different polar organizing proteins at each cell pole recruit PopA where it interacts with and mediates the function of two molecular machines: the ClpXP degradation machinery at the stalked pole and the flagellar basal body at the swarmer pole. We discovered a novel binding partner of PopA at the swarmer cell pole that together with PopA regulates the length of the flagella filament. Our work demonstrates how a second messenger provides spatiotemporal cues to change the physical behavior of an effector protein, thereby facilitating asymmetry.
Asymmetric cell division generates two daughter cells with distinct characteristics and fates. Positioning different regulatory and signaling proteins at the opposing ends of the predivisional cell produces molecularly distinct daughter cells. Here, we report a new strategy deployed by the asymmetrically dividing bacterium Caulobacter crescentus where a regulatory protein is programed to perform distinct functions at the opposing cell poles. We find that the CtrA proteolysis adaptor protein PopA assumes distinct oligomeric states at the two cell poles through asymmetrically distributed c-di-GMP: dimeric at the stalked pole and monomeric at the swarmer pole. Different polar organizing proteins at each cell pole recruit PopA where it interacts with and mediates the function of two molecular machines: the ClpXP degradation machinery at the stalked pole and the flagellar basal body at the swarmer pole. We discovered a novel binding partner of PopA at the swarmer cell pole that together with PopA regulates the length of the flagella filament. Our work demonstrates how a second messenger provides spatiotemporal cues to change the physical behavior of an effector protein, thereby facilitating asymmetry.
Jiarui Wang, Lucy Shapiro, and W. E. Moerner, "A localized adaptor protein performs distinct functions at the Caulobacter cell poles," Proc. Nat. Acad. Sci. (USA) 118, e2024705118 (2021) (DOI: 10.1073/pnas.2024705118, published online 22 March 2021).
Cryogenic Single-Molecule Fluorescence Annotations for Electron Tomography Reveal In Situ Organization of Key Proteins in Caulobacter
CIASM - for Correlated Imaging by Annotation with Single Molecules
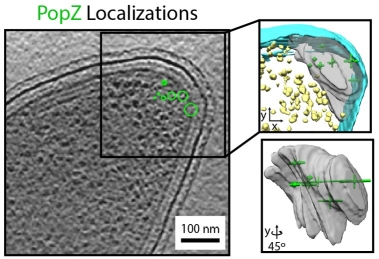 Super-resolution fluorescence microscopy and cryogenic electron tomography (CET) are powerful imaging methods for exploring the subcellular organization of biomolecules. Super-resolution fluorescence microscopy based on covalent labeling highlights specific proteins and has sufficient sensitivity to observe single fluorescent molecules, but the reconstructions lack detailed cellular context. CET has molecular-scale resolution, but lacks specific and non-perturbative intracellular labelling techniques. Here we describe an imaging scheme that correlates cryogenic single-molecule fluorescence localizations with CET reconstructions. Our approach achieves single-molecule localizations with an average lateral precision of 9 nm, and a relative registration error between the set of localizations and CET reconstruction of ~30 nm. We illustrate the workflow by annotating the positions of three proteins in the bacterium Caulobacter crescentus: McpA, PopZ, and SpmX. McpA, which forms a part of the chemoreceptor array, acts as a validation structure by being visible under both imaging modalities. In contrast, PopZ and SpmX cannot be directly identified in CET. While not directly discernable, PopZ fills a region at the cell poles that is devoid of electron-dense ribosomes. We annotate the position of PopZ with single-molecule localizations and confirm its position within the ribosome exclusion region. We further use the locations of PopZ to provide context for localizations of SpmX, a low-copy membrane associated protein sequestered by PopZ as part of a signaling pathway that leads to asymmetric cell division. Our correlative approach reveals that SpmX localizes along one side of the cell and that its extent closely matches that of the PopZ region.
Super-resolution fluorescence microscopy and cryogenic electron tomography (CET) are powerful imaging methods for exploring the subcellular organization of biomolecules. Super-resolution fluorescence microscopy based on covalent labeling highlights specific proteins and has sufficient sensitivity to observe single fluorescent molecules, but the reconstructions lack detailed cellular context. CET has molecular-scale resolution, but lacks specific and non-perturbative intracellular labelling techniques. Here we describe an imaging scheme that correlates cryogenic single-molecule fluorescence localizations with CET reconstructions. Our approach achieves single-molecule localizations with an average lateral precision of 9 nm, and a relative registration error between the set of localizations and CET reconstruction of ~30 nm. We illustrate the workflow by annotating the positions of three proteins in the bacterium Caulobacter crescentus: McpA, PopZ, and SpmX. McpA, which forms a part of the chemoreceptor array, acts as a validation structure by being visible under both imaging modalities. In contrast, PopZ and SpmX cannot be directly identified in CET. While not directly discernable, PopZ fills a region at the cell poles that is devoid of electron-dense ribosomes. We annotate the position of PopZ with single-molecule localizations and confirm its position within the ribosome exclusion region. We further use the locations of PopZ to provide context for localizations of SpmX, a low-copy membrane associated protein sequestered by PopZ as part of a signaling pathway that leads to asymmetric cell division. Our correlative approach reveals that SpmX localizes along one side of the cell and that its extent closely matches that of the PopZ region.
Peter D. Dahlberg, Saumya Saurabh, Annina M. Sartor, Jiarui Wang, Patrick Mitchell, Wah Chiu, Lucy Shapiro, W.E. Moerner, “Cryogenic Single-Molecule Fluorescence Annotations for Electron Tomography Reveal In Situ Organization of Key Proteins in Caulobacter, Proc. Nat. Acad. Sci. (USA) 117, 13937-13944 (2020) (DOI: 10.1073/pnas.2001849117, published online 8 June 2020). [![]() Slide]
Slide]
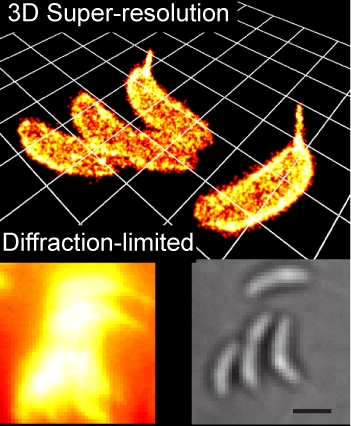
Novel fibrillar structure in the inversin compartment of primary cilia revealed by 3D single-molecule super-resolution microscopy with the DH-PSF microscope
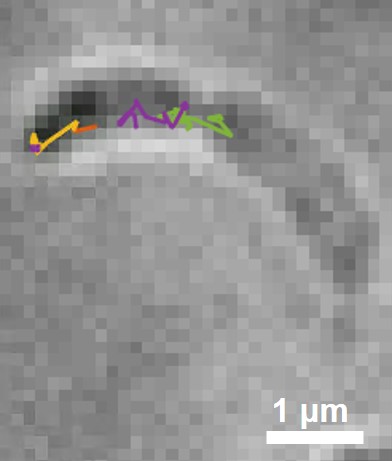
 Primary cilia in many cell types contain a peri-axonemal sub-compartment called the inversin compartment. Four proteins have been found to assemble within the inversin compartment: INVS, ANKS6, NEK8, and NPHP3. The function of the inversin compartment is unknown, but it appears to be critical for normal development including left-right asymmetry and renal tissue homeostasis. Here we combine super-resolution imaging of human RPE1 cells, a classic model for studying primary cilia in vitro, with a genetic dissection of the protein-protein binding relationships that organize compartment assembly to develop a new structural model. We observe that INVS is the core structural determinant of a compartment composed of novel fibril-like substructures, which we identify here by 3D single-molecule super-resolution imaging. We find that NEK8 and ANKS6 depend on INVS for localization to these fibrillar assemblies and that ANKS6-NEK8 density within the compartment is regulated by NEK8. Together, NEK8 and ANKS6 are required downstream of INVS to localize and concentrate NPHP3 within the compartment. In the absence of these upstream components, NPHP3 is redistributed within cilia. These results provide a more detailed structure for the inversin compartment and introduce a new example of a membraneless compartment organized by protein-protein interactions.
Primary cilia in many cell types contain a peri-axonemal sub-compartment called the inversin compartment. Four proteins have been found to assemble within the inversin compartment: INVS, ANKS6, NEK8, and NPHP3. The function of the inversin compartment is unknown, but it appears to be critical for normal development including left-right asymmetry and renal tissue homeostasis. Here we combine super-resolution imaging of human RPE1 cells, a classic model for studying primary cilia in vitro, with a genetic dissection of the protein-protein binding relationships that organize compartment assembly to develop a new structural model. We observe that INVS is the core structural determinant of a compartment composed of novel fibril-like substructures, which we identify here by 3D single-molecule super-resolution imaging. We find that NEK8 and ANKS6 depend on INVS for localization to these fibrillar assemblies and that ANKS6-NEK8 density within the compartment is regulated by NEK8. Together, NEK8 and ANKS6 are required downstream of INVS to localize and concentrate NPHP3 within the compartment. In the absence of these upstream components, NPHP3 is redistributed within cilia. These results provide a more detailed structure for the inversin compartment and introduce a new example of a membraneless compartment organized by protein-protein interactions.
Henrietta W. Bennett*, Anna-Karin Gustavsson*, Camille A. Bayas, Petar N. Petrov, Nancie Mooney, W. E. Moerner, and Peter K. Jackson, (*equal contributions), “Novel fibrillar structure in the inversin compartment of primary cilia revealed by 3D single-molecule super-resolution microscopy,” Molec. Biol. Cell 31, 619 (2020) (DOI: 10.1091/mbc.E19-09-0499, published online 2 January 2020). [![]() Slide]
Slide]
Asymmetric division yields progeny cells with distinct modes of cell cycle- dependent chromosome methylation
 The cell cycle-regulated methylation state of Caulobacter DNA mediates the temporal control of transcriptional activation of several key regulatory proteins. Restricting active CcrM DNA methyltransferase to a specific time in the cell cycle allows the maintenance of the hemi-methylated state of the chromosome during the progression of DNA replication. We determined that Caulobacter employs distinct mechanisms to control CcrM activity in the swarmer and stalked progeny cells. In progeny swarmer cells, the preferential binding of CcrM and Lon protease to a DNA platform facilitates CcrM degradation. In progeny stalked cells, CcrM is sequestered at the new cell pole. Single-molecule localization microscopy demonstrated physical anti-correlation between polarly sequestered CcrM and chromosomal DNA and Lon, thus inhibiting both DNA re-methylation and CcrM proteolysis. The distinct controls of active CcrM in the swarmer and stalked progeny cells protect the hemi-methylated state of DNA during chromosome replication, enabling robustness of cell cycle progression.
The cell cycle-regulated methylation state of Caulobacter DNA mediates the temporal control of transcriptional activation of several key regulatory proteins. Restricting active CcrM DNA methyltransferase to a specific time in the cell cycle allows the maintenance of the hemi-methylated state of the chromosome during the progression of DNA replication. We determined that Caulobacter employs distinct mechanisms to control CcrM activity in the swarmer and stalked progeny cells. In progeny swarmer cells, the preferential binding of CcrM and Lon protease to a DNA platform facilitates CcrM degradation. In progeny stalked cells, CcrM is sequestered at the new cell pole. Single-molecule localization microscopy demonstrated physical anti-correlation between polarly sequestered CcrM and chromosomal DNA and Lon, thus inhibiting both DNA re-methylation and CcrM proteolysis. The distinct controls of active CcrM in the swarmer and stalked progeny cells protect the hemi-methylated state of DNA during chromosome replication, enabling robustness of cell cycle progression.
Xiaofeng Zhou, Jiarui Wang, Jonathan Herrmann, W. E. Moerner, and Lucy Shapiro, "Asymmetric division yields progeny cells with distinct modes of regulating cell cycle-dependent chromosome methylation," Proc. Nat. Acad. Sci. (USA) 116 (31) 15661-15670 (2019) (DOI: 10.1073/pnas.1906119116, published online 17 July 2019). [![]() Slide]
Slide]
Topologically-Guided Continuous Protein Crystallization Controls Bacterial Surface Layer Self-Assembly
 Many bacteria and most archaea possess a crystalline protein surface layer (S-layer), which surrounds their growing and topologically complicated outer surface. Constructing a macromolecular structure of this scale generally requires localized enzymatic machinery, but a regulatory framework for S- layer assembly has not been identified. By labeling, superresolution imaging, and tracking the S-layer protein (SLP) from C. crescentus, we show that 2D protein self-assembly is sufficient to build and maintain the S-layer in living cells by efficient protein crystal nucleation and growth. We propose a model supported by single-molecule tracking whereby randomly secreted SLP monomers diffuse on the lipopolysaccharide (LPS) outer membrane until incorporated at the edges of growing 2D S-layer crystals. Surface topology creates crystal defects and boundaries, thereby guiding S-layer assembly. Unsupervised assembly poses challenges for therapeutics targeting S-layers. However, protein crystallization as an evolutionary driver rationalizes S-layer diversity and raises the potential for biologically inspired self-assembling macromolecular nanomaterials.
Many bacteria and most archaea possess a crystalline protein surface layer (S-layer), which surrounds their growing and topologically complicated outer surface. Constructing a macromolecular structure of this scale generally requires localized enzymatic machinery, but a regulatory framework for S- layer assembly has not been identified. By labeling, superresolution imaging, and tracking the S-layer protein (SLP) from C. crescentus, we show that 2D protein self-assembly is sufficient to build and maintain the S-layer in living cells by efficient protein crystal nucleation and growth. We propose a model supported by single-molecule tracking whereby randomly secreted SLP monomers diffuse on the lipopolysaccharide (LPS) outer membrane until incorporated at the edges of growing 2D S-layer crystals. Surface topology creates crystal defects and boundaries, thereby guiding S-layer assembly. Unsupervised assembly poses challenges for therapeutics targeting S-layers. However, protein crystallization as an evolutionary driver rationalizes S-layer diversity and raises the potential for biologically inspired self-assembling macromolecular nanomaterials.
Colin J. Comerci*, Jonathan Herrmann*, Joshua Yoon, Fatemeh Jabbarpour, Xiaofeng Zhou, John F. Nomellini, John Smit, Lucy Shapiro, Soichi Wakatsuki, and W.E. Moerner, “Topologically-Guided Continuous Protein Crystallization Controls Bacterial Surface Layer Self-Assembly,” Nature Commun. 10, 2731 (2019) (published online 21 June 2019).DOI [![]() Slide]
Slide]
Quantitative Super-Resolution Microscopy of the Mammalian Glycocalyx
 The mammalian glycocalyx is a heavily glycosylated extramembrane compartment found on nearly every cell. Despite its relevance in both health and disease, studies of the glycocalyx remain hampered by a paucity of methods to spatially classify its components. We combine metabolic labeling, bioorthogonal chemistry, and super-resolution localization microscopy to image two constituents of cell-surface glycans, N-acetylgalactosamine (GalNAc) and sialic acid, with 10–20 nm precision in 2D and 3D. This approach enables two measurements: glycocalyx height and the distribution ofindividual sugars distal from the membrane. These measurements show that the glycocalyx exhibits nanoscale organization on both cell lines and primary human tumor cells. Additionally, we observe enhanced glycocalyx height in response to epithelial-to-mesenchymal transition and to oncogenic KRAS activation. In the latter case, we trace increased height to an effector gene, GALNT7. These data highlight the power of advanced imaging methods to provide molecular and functional insights into glycocalyx biology and cancer pathways.
The mammalian glycocalyx is a heavily glycosylated extramembrane compartment found on nearly every cell. Despite its relevance in both health and disease, studies of the glycocalyx remain hampered by a paucity of methods to spatially classify its components. We combine metabolic labeling, bioorthogonal chemistry, and super-resolution localization microscopy to image two constituents of cell-surface glycans, N-acetylgalactosamine (GalNAc) and sialic acid, with 10–20 nm precision in 2D and 3D. This approach enables two measurements: glycocalyx height and the distribution ofindividual sugars distal from the membrane. These measurements show that the glycocalyx exhibits nanoscale organization on both cell lines and primary human tumor cells. Additionally, we observe enhanced glycocalyx height in response to epithelial-to-mesenchymal transition and to oncogenic KRAS activation. In the latter case, we trace increased height to an effector gene, GALNT7. These data highlight the power of advanced imaging methods to provide molecular and functional insights into glycocalyx biology and cancer pathways.
Leonhard Möckl, Kayvon Pedram, Anish R. Roy, Venkatesh Krishnan, Anna-Karin Gustavsson, Oliver Dorigo, Carolyn R. Bertozzi, and W. E. Moerner, “Quantitative Super-Resolution Microscopy of the Mammalian Glycocalyx,” Dev. Cell 49, article 4559 (2019) (DOI: 10.1016/j.devcell.2019.04.035, publication online 16 May 2019). DOI [![]() Slide]
Slide]
Revealing the nanoscale morphology of the primary cilium surface using 3D super-resolution fluorescence microscopy and surface reconstruction analysis
 Super-resolution (SR) microscopy has been used to observe structural details beyond the diffraction limit of ~250 nm in a variety of biological and materials systems. By combining this imaging technique with both computer-vision algorithms and topological methods, we reveal and quantify the nanoscale morphology of the primary cilium, a tiny tubular cellular structure (~2-6 mm long and 200-300 nm diameter). The cilium in mammalian cells protrudes out of the plasma membrane and is important in many signaling processes related to cellular differentiation and disease. After tagging individual ciliary transmembrane proteins, specifically Smoothened (SMO), with single fluorescent labels in fixed cells, we use three-dimensional (3D) single-molecule SR microscopy to determine their positions with a precision of 10-25 nm. We gain a dense, pointillistic reconstruction of the surfaces of many cilia, revealing large heterogeneity in membrane shape. A Poisson surface reconstruction (PSR) algorithm generates a fine surface mesh, allowing us to characterize the presence of deformations by quantifying the surface curvature. Upon impairment of intracellular cargo transport machinery by genetic knockout or small-molecule treatment of cells, our quantitative curvature analysis shows significant morphological differences not visible by conventional fluorescence microscopy techniques. Furthermore, using a complementary SR technique, 2-color, 2D STimulated Emission Depletion (STED) microscopy, we find that the cytoskeleton in the cilium, the axoneme, also exhibits abnormal morphology in the mutant cells, similar to our 3D results on the SMO-measured ciliary surface. Our work combines 3D SR microscopy and computational tools to quantitatively characterize morphological changes of the primary cilium under different treatments and uses STED to discover correlated changes in the underlying structure. This approach can be useful for studying other biological or nanoscale structures of interest.
Super-resolution (SR) microscopy has been used to observe structural details beyond the diffraction limit of ~250 nm in a variety of biological and materials systems. By combining this imaging technique with both computer-vision algorithms and topological methods, we reveal and quantify the nanoscale morphology of the primary cilium, a tiny tubular cellular structure (~2-6 mm long and 200-300 nm diameter). The cilium in mammalian cells protrudes out of the plasma membrane and is important in many signaling processes related to cellular differentiation and disease. After tagging individual ciliary transmembrane proteins, specifically Smoothened (SMO), with single fluorescent labels in fixed cells, we use three-dimensional (3D) single-molecule SR microscopy to determine their positions with a precision of 10-25 nm. We gain a dense, pointillistic reconstruction of the surfaces of many cilia, revealing large heterogeneity in membrane shape. A Poisson surface reconstruction (PSR) algorithm generates a fine surface mesh, allowing us to characterize the presence of deformations by quantifying the surface curvature. Upon impairment of intracellular cargo transport machinery by genetic knockout or small-molecule treatment of cells, our quantitative curvature analysis shows significant morphological differences not visible by conventional fluorescence microscopy techniques. Furthermore, using a complementary SR technique, 2-color, 2D STimulated Emission Depletion (STED) microscopy, we find that the cytoskeleton in the cilium, the axoneme, also exhibits abnormal morphology in the mutant cells, similar to our 3D results on the SMO-measured ciliary surface. Our work combines 3D SR microscopy and computational tools to quantitatively characterize morphological changes of the primary cilium under different treatments and uses STED to discover correlated changes in the underlying structure. This approach can be useful for studying other biological or nanoscale structures of interest.
Joshua Yoon, Colin J. Comerci, Lucien E. Weiss, Ljiljana Milenkovic, Tim Stearns, and W. E. Moerner, “Revealing the nanoscale morphology of the primary cilium using super-resolution fluorescence microscopy," Biophys. J. 116, 319 (2019) (DOI: 10.1016/j.bpj.2018.11.3136, published online 7 December 2018). DOI [![]() Slide] Also see COVER under publications...
Slide] Also see COVER under publications...
Identification of PAmKate as a Red Photoactivatable Fluorescent Protein for Cryogenic Super-Resolution Imaging with High Localization Precision
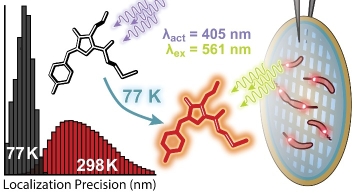 Single-molecule super-resolution fluorescence microscopy conducted in vitrified ice samples at cryogenic temperatures offers enhanced localization precision due to reduced photobleaching rates, a chemical-free and rapid fixation method, and the potential of correlation with cryogenic electron microscopy. Achieving cryogenic super-resolution microscopy requires the ability to control the sparsity of emissive labels at cryogenic temperatures. Obtaining this control presents a key challenge for the development of this technique. In this work, we identify a red photo-activatable protein, PAmKate, which remains activatable at cryogenic temperatures. We characterize its activation as a function of temperature and find that activation is efficient at cryogenic and room temperatures. We perform cryogenic super-resolution experiments in situ, labelling PopZ, a protein known to assemble into a microdomain at the poles of the model bacterium Caulobacter crescentus. We find improved localization precision at cryogenic temperatures compared to room temperature by a factor of four, attributable to reduced photobleaching. Future correlative studies will be able to use PAmKate fusions to specifically mark molecules of interest, providing a ground truth identification of a position with a precision comparable to that of cryogenic electron microscopy resolution.
Single-molecule super-resolution fluorescence microscopy conducted in vitrified ice samples at cryogenic temperatures offers enhanced localization precision due to reduced photobleaching rates, a chemical-free and rapid fixation method, and the potential of correlation with cryogenic electron microscopy. Achieving cryogenic super-resolution microscopy requires the ability to control the sparsity of emissive labels at cryogenic temperatures. Obtaining this control presents a key challenge for the development of this technique. In this work, we identify a red photo-activatable protein, PAmKate, which remains activatable at cryogenic temperatures. We characterize its activation as a function of temperature and find that activation is efficient at cryogenic and room temperatures. We perform cryogenic super-resolution experiments in situ, labelling PopZ, a protein known to assemble into a microdomain at the poles of the model bacterium Caulobacter crescentus. We find improved localization precision at cryogenic temperatures compared to room temperature by a factor of four, attributable to reduced photobleaching. Future correlative studies will be able to use PAmKate fusions to specifically mark molecules of interest, providing a ground truth identification of a position with a precision comparable to that of cryogenic electron microscopy resolution.
Peter D. Dahlberg, Annina M. Sartor, Jiarui Wang, Saumya Saurabh, Lucy Shapiro, and W. E. Moerner, “Identification of PAmKate as a Red Photoactivatable Fluorescent Protein for Cryogenic Super-Resolution Imaging, J. Amer. Chem. Soc. 140, 12310-12313 (2018) (DOI: 10.1021/jacs.8b05960, published online 17 September 2018). DOI [![]() Slide]
Slide]
Three-dimensional spatial organization and dynamics of RNase E and ribosomes in Caulobacter crescentus bacterial cells by single-particle tracking and super-resolution imaging
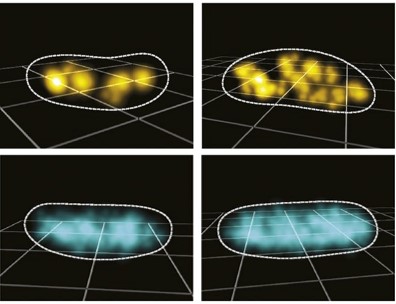 Regulated gene expression involves accurate subcellular positioning and timing of RNA synthesis, degradation, processing, and translation. RNase E is an endoribonuclease that forms the scaffold of the RNA degradosome in bacteria, responsible for the majority of mRNA turnover and RNA processing. We report the dynamic spatial organization of Caulobacter crescentus RNase E (RNA degradosome) and ribosomal protein L1 (ribosome) using 3D single particle tracking and super-resolution microscopy. RNase E formed clusters along the central axis of the cell, while weak clusters of ribosomal protein L1 were deployed throughout the cytoplasm. These results contrast with RNase E and ribosome distribution in E. coli, where RNase E co-localizes with the cytoplasmic membrane and ribosomes accumulate in polar nucleoid-free zones. For both RNase E and ribosomes in Caulobacter, we observed a decrease in confinement and clustering upon transcription inhibition and subsequent depletion of nascent RNA, suggesting that RNA substrate availability for processing, degradation, and translation facilitates confinement and clustering. Importantly, RNase E cluster positions correlated with the subcellular location of chromosomal loci of two highly transcribed ribosomal RNA genes, suggesting that RNase E’s function in ribosomal RNA processing occurs at the site of rRNA synthesis. Thus, components of the RNA degradosome and ribosome assembly are spatiotemporally organized in Caulobacter, with chromosomal readout serving as the template for this organization.
Regulated gene expression involves accurate subcellular positioning and timing of RNA synthesis, degradation, processing, and translation. RNase E is an endoribonuclease that forms the scaffold of the RNA degradosome in bacteria, responsible for the majority of mRNA turnover and RNA processing. We report the dynamic spatial organization of Caulobacter crescentus RNase E (RNA degradosome) and ribosomal protein L1 (ribosome) using 3D single particle tracking and super-resolution microscopy. RNase E formed clusters along the central axis of the cell, while weak clusters of ribosomal protein L1 were deployed throughout the cytoplasm. These results contrast with RNase E and ribosome distribution in E. coli, where RNase E co-localizes with the cytoplasmic membrane and ribosomes accumulate in polar nucleoid-free zones. For both RNase E and ribosomes in Caulobacter, we observed a decrease in confinement and clustering upon transcription inhibition and subsequent depletion of nascent RNA, suggesting that RNA substrate availability for processing, degradation, and translation facilitates confinement and clustering. Importantly, RNase E cluster positions correlated with the subcellular location of chromosomal loci of two highly transcribed ribosomal RNA genes, suggesting that RNase E’s function in ribosomal RNA processing occurs at the site of rRNA synthesis. Thus, components of the RNA degradosome and ribosome assembly are spatiotemporally organized in Caulobacter, with chromosomal readout serving as the template for this organization.
Camille A. Bayas, Jiarui Wang, Marissa K. Lee, Jared M. Schrader, Lucy Shapiro, and W. E. Moerner, “Spatial organization and dynamics of RNase E and ribosomes in Caulobacter crescentus,” Proc. Nat. Acad. Sci. (USA) 115, E3712-E3721 (2018). (DOI: 10.1073/pnas.1721648115, published online April 2, 2018). DOI [![]() Slide]
Slide]
New Labels:Super-resolution imaging of live bacteria cells using a genetically-directed, highly photostable fluoromodule based on a fluorogen-activating peptide
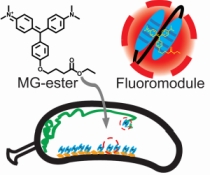 The rapid development in fluorescence microscopy and imaging techniques has greatly benefited our understanding of the mechanisms governing cellular processes at the molecular level. In particular, super-resolution microscopy methods overcome the diffraction limit to observe nanoscale cellular structures with unprecedented detail, and single-molecule tracking provides precise dynamic information about the motions of labeled proteins and oligonucleotides. Enhanced photostability of fluorescent labels (i.e., maximum emitted photons before photobleaching) is a critical requirement for achieving the ultimate spatio-temporal resolution with either method. While super-resolution imaging has greatly benefited from highly photostable fluorophores, a shortage of photostable fluorescent labels for bacteria has limited its use in these small but relevant organisms. In general, fluorescent proteins provide roughly a factor of 10 fewer emitted photons compared to small organic fluorophores. In this study, we report the use of a highly photostable fluoromodule, dL5, to genetically label proteins in the Gram-negative bacterium Caulobacter crescentus, enabling long time-scale protein tracking and super-resolution microscopy. dL5 imaging relies on the activation of the fluorogen Malachite Green (MG), and can be used to label proteins sparsely, enabling single-protein detection in live bacteria. An ester derivative of MG facilitated its passage through the bacterial cell wall/membrane. dL5-MG complexes emit two-fold more photons before photobleaching compared to organic dyes such as Cy5 and Alexa 647 in vitro; and five-fold more photons compared to eYFP in vivo. We imaged fusions of dL5 to three different proteins in live Caulobacter cells using stimulated emission depletion (STED) microscopy, yielding a four-fold resolution improvement compared to diffraction-limited imaging. Importantly, dL5 fusions to an intermediate filament protein CreS are significantly less perturbative compared to traditional fluorescent protein fusions. To the best of our knowledge, this is the first demonstration of the use of fluorogen activating proteins for super-resolution imaging in live bacterial cells.
The rapid development in fluorescence microscopy and imaging techniques has greatly benefited our understanding of the mechanisms governing cellular processes at the molecular level. In particular, super-resolution microscopy methods overcome the diffraction limit to observe nanoscale cellular structures with unprecedented detail, and single-molecule tracking provides precise dynamic information about the motions of labeled proteins and oligonucleotides. Enhanced photostability of fluorescent labels (i.e., maximum emitted photons before photobleaching) is a critical requirement for achieving the ultimate spatio-temporal resolution with either method. While super-resolution imaging has greatly benefited from highly photostable fluorophores, a shortage of photostable fluorescent labels for bacteria has limited its use in these small but relevant organisms. In general, fluorescent proteins provide roughly a factor of 10 fewer emitted photons compared to small organic fluorophores. In this study, we report the use of a highly photostable fluoromodule, dL5, to genetically label proteins in the Gram-negative bacterium Caulobacter crescentus, enabling long time-scale protein tracking and super-resolution microscopy. dL5 imaging relies on the activation of the fluorogen Malachite Green (MG), and can be used to label proteins sparsely, enabling single-protein detection in live bacteria. An ester derivative of MG facilitated its passage through the bacterial cell wall/membrane. dL5-MG complexes emit two-fold more photons before photobleaching compared to organic dyes such as Cy5 and Alexa 647 in vitro; and five-fold more photons compared to eYFP in vivo. We imaged fusions of dL5 to three different proteins in live Caulobacter cells using stimulated emission depletion (STED) microscopy, yielding a four-fold resolution improvement compared to diffraction-limited imaging. Importantly, dL5 fusions to an intermediate filament protein CreS are significantly less perturbative compared to traditional fluorescent protein fusions. To the best of our knowledge, this is the first demonstration of the use of fluorogen activating proteins for super-resolution imaging in live bacterial cells.
Saumya Saurabh, Adam M. Perez, Colin J. Comerci, Lucy Shapiro and W. E. Moerner, “Super-resolution imaging of live bacteria cells using a genetically- directed, highly photostable fluoromodule," J. Amer. Chem. Soc. 138 (33), pp 10398–10401 (2016) (DOI: 10.1021/jacs.6b05943, web publication date 1 August 2016). DOI [![]() Slide]
Slide]
Polar-PAINT: Enhanced DNA imaging using super-resolution microscopy and simultaneous single-molecule orientation measurements - sensing azimuthal angle and wobble, molecule by monecule
 Single-molecule orientation measurements provide unparalleled insight into a multitude of biological and polymeric systems. We report a simple, high-throughput technique for measuring the azimuthal orientation and rotational dynamics of single fluorescent molecules, which is compatible with localization microscopy. Our method involves modulating the polarization of an excitation laser, and analyzing the corresponding intensities emitted by single dye molecules and their modulation amplitudes. To demonstrate our approach, we use intercalating and groove-binding dyes and the PAINT approach to obtain super-resolved images of stretched DNA strands through binding-induced turn-on of fluorescence. By combining our image data with thousands of dye molecule orientation measurements, we develop a means of probing the structure of individual DNA strands, while also characterizing dye-DNA interactions. This approach may hold promise as a method for monitoring DNA conformation changes resulting from DNA-binding proteins.
Single-molecule orientation measurements provide unparalleled insight into a multitude of biological and polymeric systems. We report a simple, high-throughput technique for measuring the azimuthal orientation and rotational dynamics of single fluorescent molecules, which is compatible with localization microscopy. Our method involves modulating the polarization of an excitation laser, and analyzing the corresponding intensities emitted by single dye molecules and their modulation amplitudes. To demonstrate our approach, we use intercalating and groove-binding dyes and the PAINT approach to obtain super-resolved images of stretched DNA strands through binding-induced turn-on of fluorescence. By combining our image data with thousands of dye molecule orientation measurements, we develop a means of probing the structure of individual DNA strands, while also characterizing dye-DNA interactions. This approach may hold promise as a method for monitoring DNA conformation changes resulting from DNA-binding proteins.
Adam E. Backer, Maurice Y. Lee, and W. E. Moerner, “Enhanced DNA imaging using super-resolution microscopy and simultaneous single-molecule orientation measurements,” Optica 3, 659-666 (2016). (DOI: 10.1364/optica.3.000659, published online 17 June 2016). DOI [![]() Slide]
Slide]
Determining the Rotation Mobility of a Single Molecule from a Single Image - the M Matrix Formalism and Its Eigenvalues
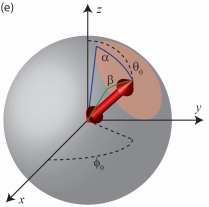 In the past decades,, measurements of the orientational freedom with which a single molecule may rotate or 'wobble' about a fixed axis have provided researchers invaluable clues about the underlying behavior of a variety of biological systems. In this paper, we propose a measurement and data analysis procedure based on a widefield fluorescence microscope image for quantitatively distinguishing individual molecules that exhibit varying degrees of rotational mobility. Our proposed technique is especially applicable to cases in which the molecule undergoes rotational motions on a timescale much faster than the framerate of the camera used to record fluorescence images. Unlike currently available methods, sophisticated hardware for modulating the polarization of light illuminating the sample is not required. Additional polarization optics may be inserted in the microscope's imaging pathway to achieve superior measurement precision, but are not essential. We present a theoretical analysis, describe the M matrix formalism of the second moments of the dipole moment and the meaning of its eigenvalues, and benchmark our technique with numerical simulations using typical experimental parameters for single-molecule imaging.
In the past decades,, measurements of the orientational freedom with which a single molecule may rotate or 'wobble' about a fixed axis have provided researchers invaluable clues about the underlying behavior of a variety of biological systems. In this paper, we propose a measurement and data analysis procedure based on a widefield fluorescence microscope image for quantitatively distinguishing individual molecules that exhibit varying degrees of rotational mobility. Our proposed technique is especially applicable to cases in which the molecule undergoes rotational motions on a timescale much faster than the framerate of the camera used to record fluorescence images. Unlike currently available methods, sophisticated hardware for modulating the polarization of light illuminating the sample is not required. Additional polarization optics may be inserted in the microscope's imaging pathway to achieve superior measurement precision, but are not essential. We present a theoretical analysis, describe the M matrix formalism of the second moments of the dipole moment and the meaning of its eigenvalues, and benchmark our technique with numerical simulations using typical experimental parameters for single-molecule imaging.
Adam S. Backer and W. E. Moerner, “Determining the rotational mobility of a single molecule from a single image: a numerical study,” Optics Express 23, 4255-4276 (2015). (DOI: 10.1364/OE.23.004255, published online February 11, 2015). DOI [![]() Slide]
Slide]
Extending Single-Molecule Microscopy by Optical Fourier Processing: Modulating the Electric Field of the Molecular Emission in the Back Focal Plane
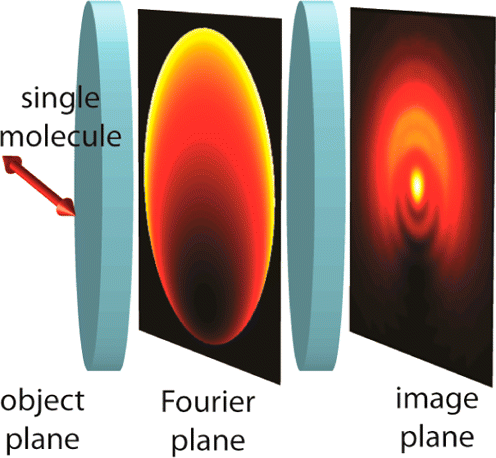 This article surveys the recent application of optical Fourier processing to the long-established but still expanding field of single-molecule imaging and microscopy. A variety of single-molecule studies can benefit from the additional image information that can be obtained by modulating the Fourier, or pupil, plane of a wide-field microscope. After briefly reviewing several current applications, we present a comprehensive and computationally efficient theoretical model for simulating single-molecule fluorescence as it propagates through an imaging system. Furthermore, we describe how phase/amplitude-modulating optics inserted in the imaging pathway may be modeled, especially at the Fourier plane. Effectively, the propagating electric field of the light from the molecule in the back focal plane can be forced to give up additional information about each molecule by judicious choice of optics.Finally, we discuss selected recent applications of Fourier processing methods to measure the orientation, depth, and rotational mobility of single fluorescent molecules.
This article surveys the recent application of optical Fourier processing to the long-established but still expanding field of single-molecule imaging and microscopy. A variety of single-molecule studies can benefit from the additional image information that can be obtained by modulating the Fourier, or pupil, plane of a wide-field microscope. After briefly reviewing several current applications, we present a comprehensive and computationally efficient theoretical model for simulating single-molecule fluorescence as it propagates through an imaging system. Furthermore, we describe how phase/amplitude-modulating optics inserted in the imaging pathway may be modeled, especially at the Fourier plane. Effectively, the propagating electric field of the light from the molecule in the back focal plane can be forced to give up additional information about each molecule by judicious choice of optics.Finally, we discuss selected recent applications of Fourier processing methods to measure the orientation, depth, and rotational mobility of single fluorescent molecules.
Adam S. Backer and W. E. Moerner, “Extending Single-Molecule Microscopy Using Optical Fourier Processing,” James Skinner Festschrift, J. Phys. Chem. B 118, 8313-8329 (2014) (DOI: 10.1021/jp501778z, published online 18 April 2014). DOI [![]() Slide]
Slide]
Small-Molecule Labeling of Live Cell Surfaces for Three-Dimensional Super-Resolution Microscopy
 Precise imaging of the cell surface of fluorescently labeled bacteria requires super-resolution methods because the size-scale of these cells is on the order of the diffraction limit. In this work, we present a photocontrollable small-molecule rhodamine spirolactam emitter suitable for non-toxic and specific labeling of the outer surface of cells for three-dimensional (3D) super-resolution (SR) imaging. Conventional rhodamine spirolactams photoswitch to the emitting form with UV light; however, these wavelengths can damage cells. We extended photoswitching to visible wavelengths >400 nm by iterative synthesis and spectroscopic characterization optimizing the substitution on the spirolactam. Further, an N-hydroxy-succinimide functionalized derivative enabled covalent labeling of amines on the surface of live Caulobacter crescentus cells. Resulting 3D SR reconstructions of the labeled cell surface reveal uniform and specific sampling with thousands of localizations per cell and excellent localization precision in x, y, and z. The distribution of cell stalk lengths (a sub-diffraction-sized cellular structure) was quantified for a mixed population of cells. Pulse-chase experiments identified sites of cell surface growth. Covalent labeling with the optimized rhodamine spirolactam label provides a general strategy to study the surfaces of living cells with high specificity and resolution down to 10-20 nm.
Precise imaging of the cell surface of fluorescently labeled bacteria requires super-resolution methods because the size-scale of these cells is on the order of the diffraction limit. In this work, we present a photocontrollable small-molecule rhodamine spirolactam emitter suitable for non-toxic and specific labeling of the outer surface of cells for three-dimensional (3D) super-resolution (SR) imaging. Conventional rhodamine spirolactams photoswitch to the emitting form with UV light; however, these wavelengths can damage cells. We extended photoswitching to visible wavelengths >400 nm by iterative synthesis and spectroscopic characterization optimizing the substitution on the spirolactam. Further, an N-hydroxy-succinimide functionalized derivative enabled covalent labeling of amines on the surface of live Caulobacter crescentus cells. Resulting 3D SR reconstructions of the labeled cell surface reveal uniform and specific sampling with thousands of localizations per cell and excellent localization precision in x, y, and z. The distribution of cell stalk lengths (a sub-diffraction-sized cellular structure) was quantified for a mixed population of cells. Pulse-chase experiments identified sites of cell surface growth. Covalent labeling with the optimized rhodamine spirolactam label provides a general strategy to study the surfaces of living cells with high specificity and resolution down to 10-20 nm.
Marissa K. Lee, Prabin Rai, Jarrod Williams, Robert J. Twieg, and W. E. Moerner, “Small-Molecule Labeling of Live Cell Surfaces for Three-Dimensional Super-Resolution Microscopy,” J. Amer. Chem. Soc. . 136, 14003-14006 (2014) (DOI: 10.1021/ja508028h), published online, September 15, 2014. DOI [![]() Slide]
Slide]
The PopZ bacterial scaffold in Caulobacter directs pole-specific centromere segregation
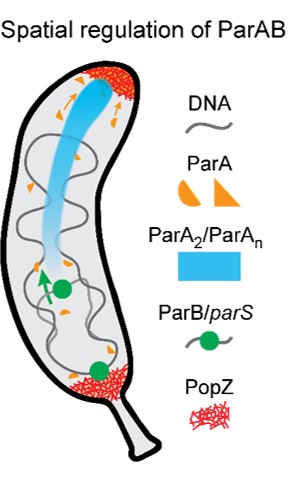 Bacteria use partitioning systems based on the ParA ATPase to actively mobilize and spatially organize molecular cargoes throughout the cytoplasm. The bacterium Caulobacter crescentus uses a ParA-based partitioning system to segregate newly replicated chromosomal centromeres to opposite cell poles. Here we demonstrate that the Caulobacter PopZ scaffold creates an organizing center at the cell pole that actively regulates polar centromere transport by the ParA partition system. As segregation proceeds, the ParB-bound centromere complex is moved by progressively disassembling ParA from a nucleoid-bound structure. Using superresolution microscopy, we show that released ParA is recruited directly to binding sites within a 3D ultrastructure composed of PopZ at the cell pole, whereas the ParB-centromere complex remains at the periphery of the PopZ structure. PopZ recruitment of ParA stimulates ParA to assemble on the nucleoid near the PopZ-proximal cell pole.We identify mutations in PopZ that allow scaffold assembly but specifically abrogate interactions with ParA and demonstrate that PopZ/ParA interactions are required for proper chromosome segregation in vivo. We propose that during segregation PopZ sequesters free ParA and induces target-proximal regeneration of ParA DNA binding activity to enforce processive and pole-directed centromere segregation, preventing segregation reversals. PopZ therefore functions as a polar hub complex at the cell pole to directly regulate the directionality and destination of transfer of the mitotic segregation machine.
Bacteria use partitioning systems based on the ParA ATPase to actively mobilize and spatially organize molecular cargoes throughout the cytoplasm. The bacterium Caulobacter crescentus uses a ParA-based partitioning system to segregate newly replicated chromosomal centromeres to opposite cell poles. Here we demonstrate that the Caulobacter PopZ scaffold creates an organizing center at the cell pole that actively regulates polar centromere transport by the ParA partition system. As segregation proceeds, the ParB-bound centromere complex is moved by progressively disassembling ParA from a nucleoid-bound structure. Using superresolution microscopy, we show that released ParA is recruited directly to binding sites within a 3D ultrastructure composed of PopZ at the cell pole, whereas the ParB-centromere complex remains at the periphery of the PopZ structure. PopZ recruitment of ParA stimulates ParA to assemble on the nucleoid near the PopZ-proximal cell pole.We identify mutations in PopZ that allow scaffold assembly but specifically abrogate interactions with ParA and demonstrate that PopZ/ParA interactions are required for proper chromosome segregation in vivo. We propose that during segregation PopZ sequesters free ParA and induces target-proximal regeneration of ParA DNA binding activity to enforce processive and pole-directed centromere segregation, preventing segregation reversals. PopZ therefore functions as a polar hub complex at the cell pole to directly regulate the directionality and destination of transfer of the mitotic segregation machine.
Jerod L. Ptacin, Andreas Gahlmann, Grant R. Bowman, Adam M. Perez, Alexander R. S. von Diezmann, Michael R. Eckart, W. E. Moerner, and Lucy Shapiro, “Bacterial scaffold directs pole-specific centromere segregation,” Proc. Nat. Acad. Sci. (USA) 111, E2046-E2055 (2014), published online 28 April 2014. DOI [![]() Slide]
Slide]
STED Microscopy: Cby1 promotes Ahi1 recruitment to a ring-shaped domain at the centriole-cilium interface and facilitates proper cilium formation and function.
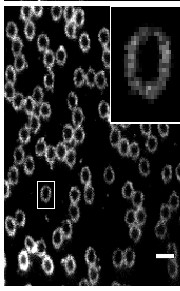 Defects in centrosome and cilium function are associated with phenotypically related syndromes called ciliopathies. Cby1, the mammalian orthologue of the Drosophila Chibby protein, localizes to mature centrioles, is important for ciliogenesis in multiciliated airway epithelia in mice, and antagonizes canonical Wnt signaling via direct regulation of β-catenin. We report that deletion of the mouse Cby1 gene results in cystic kidneys, a phenotype common to ciliopathies, and that Cby1 facilitates the formation of primary cilia and ciliary recruitment of the Joubert syndrome protein Arl13b. Localization of Cby1 to the distal end of mature centrioles depends on the centriole protein Ofd1. Superresolution microscopy using both three-dimensional SIM and STED reveals that Cby1 localizes to an ∼250-nm ring at the distal end of the mature centriole, in close proximity to Ofd1 and Ahi1, a component of the transition zone between centriole and cilium. The amount of centriole-localized Ahi1, but not Ofd1, is reduced in Cby1−/− cells. This suggests that Cby1 is required for efficient recruitment of Ahi1, providing a possible molecular mechanism for the ciliogenesis defect in Cby1−/− cells.
Defects in centrosome and cilium function are associated with phenotypically related syndromes called ciliopathies. Cby1, the mammalian orthologue of the Drosophila Chibby protein, localizes to mature centrioles, is important for ciliogenesis in multiciliated airway epithelia in mice, and antagonizes canonical Wnt signaling via direct regulation of β-catenin. We report that deletion of the mouse Cby1 gene results in cystic kidneys, a phenotype common to ciliopathies, and that Cby1 facilitates the formation of primary cilia and ciliary recruitment of the Joubert syndrome protein Arl13b. Localization of Cby1 to the distal end of mature centrioles depends on the centriole protein Ofd1. Superresolution microscopy using both three-dimensional SIM and STED reveals that Cby1 localizes to an ∼250-nm ring at the distal end of the mature centriole, in close proximity to Ofd1 and Ahi1, a component of the transition zone between centriole and cilium. The amount of centriole-localized Ahi1, but not Ofd1, is reduced in Cby1−/− cells. This suggests that Cby1 is required for efficient recruitment of Ahi1, providing a possible molecular mechanism for the ciliogenesis defect in Cby1−/− cells.
Yin Loon Lee, Joshua Santé, Colin J. Comerci, Benjamin Cyge, Luis F. Menezes, Feng-Qian Li, Gregory G. Germino, W. E. Moerner, Ken-Ichi Takemaru, and Tim Stearns, “Cby1 promotes Ahi1 recruitment to a ring-shaped domain at the centriole–cilium interface and facilitates proper cilium formation and function,” Mol. Biol. Cell 25 (19) 2919-2933 (2014), published online August 7, 2014. DOI [![]() Slide]
Slide]
Quantitative multicolor subdiffraction imaging of bacterial protein ultrastructures in 3D
 We demonstrate quantitative multicolor 3D subdiffraction imaging of the structural arrangement of fluorescent protein fusions in living Caulobacter crescentus bacteria. Given single-molecule localization precisions of 20-40 nm, a flexible locally-weighted image registration algorithm is critical to accurately combine the super-resolution data with <10 nm error. Simple surface-relief dielectric phase masks implement a double-helix response at two wavelengths to distinguish two different fluorescent labels and to quantitatively and precisely localize them relative to each other in 3D. This work demonstrates that the DH-PSF approach to 3D localization can easily be implemented with minimal modification to a conventional fluorescence microscope.
We demonstrate quantitative multicolor 3D subdiffraction imaging of the structural arrangement of fluorescent protein fusions in living Caulobacter crescentus bacteria. Given single-molecule localization precisions of 20-40 nm, a flexible locally-weighted image registration algorithm is critical to accurately combine the super-resolution data with <10 nm error. Simple surface-relief dielectric phase masks implement a double-helix response at two wavelengths to distinguish two different fluorescent labels and to quantitatively and precisely localize them relative to each other in 3D. This work demonstrates that the DH-PSF approach to 3D localization can easily be implemented with minimal modification to a conventional fluorescence microscope.
Andreas Gahlmann, Jerod L. Ptacin, Ginni Grover, Sean Quirin, Alexander R. S. von Diezmann, Marissa K. Lee, Mikael P. Backlund, Lucy Shapiro, Rafael Piestun, and W. E. Moerner, “Quantitative Multicolor Subdiffraction Imaging of Bacterial Protein Ultrastructures in Three Dimensions,” Nano Lett. 13, 987-993 (2013), published online February 15, 2013. DOI [![]() Slide]
Slide]
Enzymatic Activation of Nitro-Aryl Fluorogens in Live Bacteria for Enzymatic Turnover-Activated Localization Microscopy Beyond the Diffraction Limit
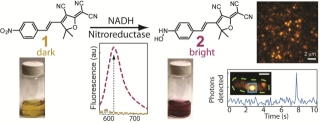 Many modern super-resolution imaging methods based on single-molecule fluorescence require the conversion of a dark fluorogen into a bright emitter to control emitter concentration. We have synthesized and characterized a nitro-aryl fluorogen which can be converted by a nitroreductase enzyme into a bright push-pull red-emitting fluorophore. Synthesis of model compounds and optical spectroscopy identify a hydroxyl-amino derivative as the product fluorophore, which is bright and detectable on the single-molecule level for fluorogens attached to a surface. Solution kinetic analysis shows Michaelis-Menten rate dependence upon both NADH and the fluorogen concentrations as expected. The generation of low concentrations of single-molecule emitters by enzymatic turnovers is used to extract subdiffraction information about localizations of both fluorophores and nitroreductase enzymes in cells. Enzymatic Turnover Activated Localization Microscopy (ETALM) is a complementary mechanism to photoactivation and blinking for controlling the emission of single molecules to image beyond the diffraction limit.
Many modern super-resolution imaging methods based on single-molecule fluorescence require the conversion of a dark fluorogen into a bright emitter to control emitter concentration. We have synthesized and characterized a nitro-aryl fluorogen which can be converted by a nitroreductase enzyme into a bright push-pull red-emitting fluorophore. Synthesis of model compounds and optical spectroscopy identify a hydroxyl-amino derivative as the product fluorophore, which is bright and detectable on the single-molecule level for fluorogens attached to a surface. Solution kinetic analysis shows Michaelis-Menten rate dependence upon both NADH and the fluorogen concentrations as expected. The generation of low concentrations of single-molecule emitters by enzymatic turnovers is used to extract subdiffraction information about localizations of both fluorophores and nitroreductase enzymes in cells. Enzymatic Turnover Activated Localization Microscopy (ETALM) is a complementary mechanism to photoactivation and blinking for controlling the emission of single molecules to image beyond the diffraction limit.
Marissa K. Lee, Jarrod Williams, Robert J. Twieg, Jianghong Rao, and W. E. Moerner, “Enzymatic Activation of Nitro-Aryl Fluorogens in Live Bacterial Cells for Enzymatic Turnover-Activated Localization Microscopy,” Chemical Science4 (1), 220-225 (2013), published online 5 October 2012. DOI [![]() Slide]
Slide]
Fluorescent Saxitoxins for Live Cell Imaging of Single Voltage-Gated Sodium Ion Channels Beyond the Optical Diffraction Limit
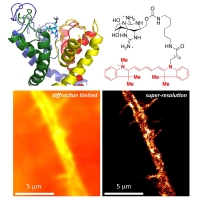 A desire to better understand the role of voltage-gated sodium channels (NaVs) in signal conduction and their dysregulation in specific disease states motivates the development of high precision tools for their study. Nature has evolved a collection of small molecule agents, including the shellfish poison (+)-saxitoxin, that bind to the extracellular pore of select NaV isoforms. As described in this report, de novo chemical synthesis has enabled the preparation of fluorescently labeled derivatives of (+)-saxitoxin, STX-Cy5 and STX-DCDHF, which display reversible binding to NaVs in live cells. Electrophysiology and confocal fluorescence microscopy studies confirm that these STX-based dyes function as potent and selective NaV labels. The utility of these probes is underscored in single-molecule and super-resolution imaging experiments, which reveal NaV distributions well beyond the optical diffraction limit in subcellular features such as neuritic spines and filopodia.
A desire to better understand the role of voltage-gated sodium channels (NaVs) in signal conduction and their dysregulation in specific disease states motivates the development of high precision tools for their study. Nature has evolved a collection of small molecule agents, including the shellfish poison (+)-saxitoxin, that bind to the extracellular pore of select NaV isoforms. As described in this report, de novo chemical synthesis has enabled the preparation of fluorescently labeled derivatives of (+)-saxitoxin, STX-Cy5 and STX-DCDHF, which display reversible binding to NaVs in live cells. Electrophysiology and confocal fluorescence microscopy studies confirm that these STX-based dyes function as potent and selective NaV labels. The utility of these probes is underscored in single-molecule and super-resolution imaging experiments, which reveal NaV distributions well beyond the optical diffraction limit in subcellular features such as neuritic spines and filopodia.
Alison E. Ondrus*, Hsiao-lu D. Lee*, Shigeki Iwanaga, William H. Parsons, Brian M. Andresen, W. E. Moerner, and J. Du Bois (equal contributions), “Fluorescent Saxitoxins for Live Cell Imaging of Single Voltage-Gated Sodium Ion Channels Beyond the Optical Diffraction Limit,” Chemistry and Biology 19, 902-912 (2012), published online 26 July 2012. (*equal contributions) DOI [![]() Slide]
Slide]
STED Microscopy with Optimized Labeling Density Reveals 9-Fold Arrangement of a Centriole Protein
 Super-resolution fluorescence microscopy can achieve resolution beyond the optical diffraction limit, partially closing the gap between conventional optical imaging and electron microscopy for elucidation of subcellular architecture. The centriole, a key component of the cellular control and division machinery, is 250 nm in diameter, a spatial scale where super-resolution methods such as stimulated emission depletion (STED) microscopy can provide previously unobtainable detail. We use STED with resolution of 60 nm to demonstrate that the centriole distal appendage protein Cep164 localizes in 9 clusters spaced around a ring of ~300 nm in diameter, and quantify the influence of the labeling density in STED immunofluorescence microscopy. We find that the labeling density dramatically influences the observed number, size and brightness of labeled Cep164 clusters, and estimate the average number of secondary antibody labels per cluster. The arrangements are morphologically similar in centrioles of both proliferating cells and differentiated multiciliated cells, suggesting a relationship of this structure to function. Our STED measurements in single centrioles are consistent with results obtained by electron microscopy, which involve ensemble averaging or very different sample preparation conditions, suggesting that we have arrived at a direct measurement of a centriole protein by careful optimization of the labeling density.
Super-resolution fluorescence microscopy can achieve resolution beyond the optical diffraction limit, partially closing the gap between conventional optical imaging and electron microscopy for elucidation of subcellular architecture. The centriole, a key component of the cellular control and division machinery, is 250 nm in diameter, a spatial scale where super-resolution methods such as stimulated emission depletion (STED) microscopy can provide previously unobtainable detail. We use STED with resolution of 60 nm to demonstrate that the centriole distal appendage protein Cep164 localizes in 9 clusters spaced around a ring of ~300 nm in diameter, and quantify the influence of the labeling density in STED immunofluorescence microscopy. We find that the labeling density dramatically influences the observed number, size and brightness of labeled Cep164 clusters, and estimate the average number of secondary antibody labels per cluster. The arrangements are morphologically similar in centrioles of both proliferating cells and differentiated multiciliated cells, suggesting a relationship of this structure to function. Our STED measurements in single centrioles are consistent with results obtained by electron microscopy, which involve ensemble averaging or very different sample preparation conditions, suggesting that we have arrived at a direct measurement of a centriole protein by careful optimization of the labeling density.
Lana Lau, Yin Loon Lee, Steffen J. Sahl, Tim Stearns, and W. E. Moerner, “STED Microscopy with Optimized Labeling Density Reveals 9-fold Arrangement of a Centriole Protein,” Biophys. J. 102, 2926-2935 (2012) published online 19 June 2012. DOI [![]() Slide]
Slide]
Super-Resolution Imaging of the Nucleoid-Associated Protein HU in Caulobacter crescentus
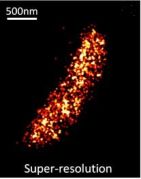 Little is known about the structure and function of most nucleoid-associated proteins (NAP) in bacteria. One reason for this is that the distribution and structure of the proteins is obfuscated by the diffraction limit in standard widefield and confocal fluorescence imaging. In particular, the distribution of HU, which is the most abundant NAP, has received little attention. In this study we investigate the distribution of HU in Caulobacter crescentus using a combination of super-resolution (SR) fluorescence imaging and spatial point statistics. By simply increasing the laser power, single molecules of the fluorescent protein fusion HU2-eYFP can be made to blink on and off to achieve SR imaging with a single excitation source. Through quantification by Ripley’s K-test and comparison with Monte Carlo simulations, we find the protein is slightly clustered within a mostly uniform distribution throughout the swarmer and stalked stages of the cell cycle but more highly clustered in pre-divisional cells. The methods presented in this paper should be of broad applicability in the future study of prokaryotic NAPs.
Little is known about the structure and function of most nucleoid-associated proteins (NAP) in bacteria. One reason for this is that the distribution and structure of the proteins is obfuscated by the diffraction limit in standard widefield and confocal fluorescence imaging. In particular, the distribution of HU, which is the most abundant NAP, has received little attention. In this study we investigate the distribution of HU in Caulobacter crescentus using a combination of super-resolution (SR) fluorescence imaging and spatial point statistics. By simply increasing the laser power, single molecules of the fluorescent protein fusion HU2-eYFP can be made to blink on and off to achieve SR imaging with a single excitation source. Through quantification by Ripley’s K-test and comparison with Monte Carlo simulations, we find the protein is slightly clustered within a mostly uniform distribution throughout the swarmer and stalked stages of the cell cycle but more highly clustered in pre-divisional cells. The methods presented in this paper should be of broad applicability in the future study of prokaryotic NAPs.
Steven F. Lee*, Michael A. Thompson*, Monica Schwartz, Lucy Shapiro, and W. E. Moerner, “Super-Resolution Imaging of the Nucleoid-Associated Protein HU in Caulobacter crescentus," Biophys. J. Lett. 100, L31-L33 (2011). SIandMovies, DOI [![]() Slide]
Slide]
A New Photoactivatable Organic Fluorophore Enables Superresolution Imaging of Targeted Proteins in Fixed and Living Cells
 Superresolution imaging techniques based on sequential imaging of sparse subsets of single molecules require fluorophores whose emission can be photoactivated or photoswitched. Because typical organic fluorophores can emit significantly more photons than average fluorescent proteins, organic fluorophores have a potential advantage in super-resolution imaging schemes, but targeting to specific cellular proteins must be provided. We report the design and application of HaloTag-based target-specific azido DCDHFs, a class of photoactivatable push–pull fluorogens which produce bright fluorescent labels suitable for single-molecule superresolution imaging in live bacterial and fixed mammalian cells.
Superresolution imaging techniques based on sequential imaging of sparse subsets of single molecules require fluorophores whose emission can be photoactivated or photoswitched. Because typical organic fluorophores can emit significantly more photons than average fluorescent proteins, organic fluorophores have a potential advantage in super-resolution imaging schemes, but targeting to specific cellular proteins must be provided. We report the design and application of HaloTag-based target-specific azido DCDHFs, a class of photoactivatable push–pull fluorogens which produce bright fluorescent labels suitable for single-molecule superresolution imaging in live bacterial and fixed mammalian cells.
Hsiao-lu D. Lee, Samuel J. Lord, Shigeki Iwanaga, Ke Zhan, Hexin Xie, Jarrod C. Williams, Hui Wang, Grant R. Bowman, Erin D. Goley, Lucy Shapiro, Robert J. Twieg, Jianghong Rao, and W. E. Moerner, “Superresolution Imaging of Targeted Proteins in Fixed and Living Cells Using Photoactivatable Organic Fluorophores," J. Amer. Chem. Soc. 132, 15099-15101 (2010), published online October 11, 2010. [![]() Slide] [journal link]
Slide] [journal link]
A Spindle-like Apparatus Guides Bacterial Chromosome Separation
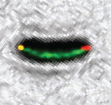 Until recently, a dedicated mitotic apparatus that segregates newly replicated chromosomes into daughter cells was believed to be unique to eukaryotic cells. Here we demonstrate that the bacterium Caulobacter crescentus segregates its chromosome using a partitioning (Par) apparatus that has surprising similarities to eukaryotic spindles. We show that the C. crescentus ATPase ParA forms linear polymers in vitro and assembles into a narrow linear structure in vivo detected by superresolution imaging. The centromere-binding protein ParB binds to and destabilizes ParA structures in vitro. We propose that this ParB-stimulated ParA depolymerization activity moves the centromere to the opposite cell pole through a burnt bridge Brownian ratchet mechanism. Finally, we identify the pole-specific TipN protein1, 2 as a new component of the Par system that is required to maintain the directionality of DNA transfer towards the new cell pole. Our results elucidate a bacterial chromosome segregation mechanism that features basic operating principles similar to eukaryotic mitotic machines, including a multivalent protein complex at the centromere that stimulates the dynamic disassembly of polymers to move chromosomes into daughter compartments.
Until recently, a dedicated mitotic apparatus that segregates newly replicated chromosomes into daughter cells was believed to be unique to eukaryotic cells. Here we demonstrate that the bacterium Caulobacter crescentus segregates its chromosome using a partitioning (Par) apparatus that has surprising similarities to eukaryotic spindles. We show that the C. crescentus ATPase ParA forms linear polymers in vitro and assembles into a narrow linear structure in vivo detected by superresolution imaging. The centromere-binding protein ParB binds to and destabilizes ParA structures in vitro. We propose that this ParB-stimulated ParA depolymerization activity moves the centromere to the opposite cell pole through a burnt bridge Brownian ratchet mechanism. Finally, we identify the pole-specific TipN protein1, 2 as a new component of the Par system that is required to maintain the directionality of DNA transfer towards the new cell pole. Our results elucidate a bacterial chromosome segregation mechanism that features basic operating principles similar to eukaryotic mitotic machines, including a multivalent protein complex at the centromere that stimulates the dynamic disassembly of polymers to move chromosomes into daughter compartments.
Jerod L. Ptacin, Steven F. Lee, Ethan C. Garner, Esteban Toro, Michael Eckart, Luis R. Comolli, W.E. Moerner, and Lucy Shapiro, “A spindle-like apparatus guides bacterial chromosome segregation,” Nature Cell Biology 12, 791-798 (2010), published online July 25, 2010. [![]() Slide] [journal link]
Slide] [journal link]
Photoactivatable Push-Pull Fluorophores Form a General Class and Enable Fluorogenic Photoaffinity Labeling
 Dark azido push−pull chromophores have the ability to be photoactivated to produce bright fluorescent labels suitable for single-molecule imaging. Upon illumination, the aryl azide functionality in the fluorogens participates in a photochemical conversion to an aryl amine, thus restoring charge-transfer absorption and fluorescence. Previously, we reported that one compound, DCDHF-V-P-azide, was photoactivatable. Here, we demonstrate that the azide-to-amine photoactivation process is generally applicable to a variety of push−pull chromophores, and we characterize the photophysical parameters including photoconversion quantum yield, photostability, and turn-on ratio. Azido push−pull fluorogens provide a new class of photoactivatable single-molecule probes for fluorescent labeling and super-resolution microscopy. Lastly, we demonstrate that photoactivated push−pull dyes can insert into bonds of nearby biomolecules, simultaneously forming a covalent bond and becoming fluorescent (fluorogenic photoaffinity labeling).
Dark azido push−pull chromophores have the ability to be photoactivated to produce bright fluorescent labels suitable for single-molecule imaging. Upon illumination, the aryl azide functionality in the fluorogens participates in a photochemical conversion to an aryl amine, thus restoring charge-transfer absorption and fluorescence. Previously, we reported that one compound, DCDHF-V-P-azide, was photoactivatable. Here, we demonstrate that the azide-to-amine photoactivation process is generally applicable to a variety of push−pull chromophores, and we characterize the photophysical parameters including photoconversion quantum yield, photostability, and turn-on ratio. Azido push−pull fluorogens provide a new class of photoactivatable single-molecule probes for fluorescent labeling and super-resolution microscopy. Lastly, we demonstrate that photoactivated push−pull dyes can insert into bonds of nearby biomolecules, simultaneously forming a covalent bond and becoming fluorescent (fluorogenic photoaffinity labeling).
S. J. Lord, H-L. D. Lee, R. Samuel, R. Weber, N. Liu, N. R. Conley, M. A. Thompson, R. J. Twieg, and W. E. Moerner, “Azido Push–Pull Fluorogens Photoactivate to Produce Bright Fluorescent Labels,” appearing in J. Phys. Chem. B (Michael R. Wasielewski Festschrift), published online October 27, 2009, doi: 10.1021/jp907080r. [journal link]
Superresolution Imaging in Live C. Crescentus Cells Using Photoswitchable EYFP
 Most SMACM experiments rely on specialized photoactivatable or photoswitchable fluorescent protein labels. In our lab, however, we have shown that this is not an imperative. Rather, we have used fusions to the common fluorescent protein EYFP to perform in vivo superresolution imaging in live bacteria. We also take advantage of the fact that, rather than being photoactivated, EYFP can be reactivated with violet light after apparent photobleaching.3 To address limitations arising from physiologically imposed upper boundaries on the concentration of fluorophores, we employ dark time-lapse periods to allow single-molecule motions to fill in filamentous structures, increasing the effective labeling concentration while localizing each emitter at most once per resolution-limited spot. We image cell-cycle-dependent superstructures of the bacterial actin protein MreB in live C. crescentus cells3 with sub-40-nm resolution for the first time, showing that EYFP is a useful emitter for in vivo superresolution imaging of intracellular structures in bacterial cells.
Most SMACM experiments rely on specialized photoactivatable or photoswitchable fluorescent protein labels. In our lab, however, we have shown that this is not an imperative. Rather, we have used fusions to the common fluorescent protein EYFP to perform in vivo superresolution imaging in live bacteria. We also take advantage of the fact that, rather than being photoactivated, EYFP can be reactivated with violet light after apparent photobleaching.3 To address limitations arising from physiologically imposed upper boundaries on the concentration of fluorophores, we employ dark time-lapse periods to allow single-molecule motions to fill in filamentous structures, increasing the effective labeling concentration while localizing each emitter at most once per resolution-limited spot. We image cell-cycle-dependent superstructures of the bacterial actin protein MreB in live C. crescentus cells3 with sub-40-nm resolution for the first time, showing that EYFP is a useful emitter for in vivo superresolution imaging of intracellular structures in bacterial cells.
J.S. Biteen, M.A. Thompson, N.K. Tselentis, G.R. Bowman, L. Shapiro, and W.E. Moerner, Nature Methods 5, 947 (15 Sept 2008) [![]() Slide] [journal link: Nature Meth.]
Slide] [journal link: Nature Meth.]
New Photoactivatable Single-Molecule Fluorophores
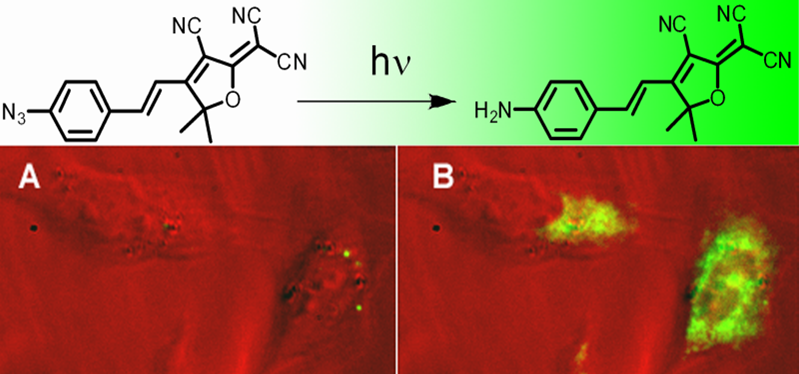 For several years, we have been exploring the properties of push-pull fluorophores containing an amine donor covalently linked to a dicyanomethylenedihydrofuran (DCDHF) acceptor, described elsewhere on these pages. In this new effort, we created a new class of photoactivatable single-molecule fluorophores by replacing the amine with an azide (A). With long-wavelength pumping at 594 nm, the azido-DCDHF fluorogenic molecules are dark, but applying low-intensity activating light at 407nm converts the azide to an amine (B), restoring the donor-acceptor character, the redshifted absorption, and the bright fluorescent emission. Because the emitters are not specifically targeted in this preliminary study, the fluorophores either diffuse in the cytosol, or attach to relatively immobile proteins via insertion into C-C bonds.
For several years, we have been exploring the properties of push-pull fluorophores containing an amine donor covalently linked to a dicyanomethylenedihydrofuran (DCDHF) acceptor, described elsewhere on these pages. In this new effort, we created a new class of photoactivatable single-molecule fluorophores by replacing the amine with an azide (A). With long-wavelength pumping at 594 nm, the azido-DCDHF fluorogenic molecules are dark, but applying low-intensity activating light at 407nm converts the azide to an amine (B), restoring the donor-acceptor character, the redshifted absorption, and the bright fluorescent emission. Because the emitters are not specifically targeted in this preliminary study, the fluorophores either diffuse in the cytosol, or attach to relatively immobile proteins via insertion into C-C bonds.
S. J. Lord, N. R. Conley, H.-l. D. Lee, R. Samuel, N. Liu, R. J. Twieg, W. E. Moerner, JACS 130, 9204 (2008) [![]() Slide] [journal link: JACS]
Slide] [journal link: JACS]
Cy3-Cy5 Covalent Heterodimers for Superresolution Imaging
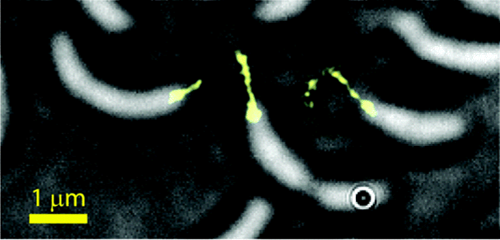 Covalent heterodimers of the Cy3 and Cy5 fluorophores have been prepared from commercially available starting materials and characterized at the single-molecule level. This system behaves as a discrete molecular photoswitch, in which photoexcitation of the Cy5 results in fluorescence emission or, with a much lower probability, causes the Cy5 to enter into a long-lived, but metastable, dark state. Photoinduced recovery of the emissive Cy5 is achieved by very low intensity excitation (5 W/cm2) of the Cy3 fluorophore at a shorter wavelength. A similar system consisting of proximal, but not covalently linked, Cy3 and Cy5 has found application in stochastic optical reconstruction microscopy (STORM), a single-molecule localization-based technique for superresolution imaging that requires photoswitching. The covalent Cy3-Cy5 heterodimers described herein eliminate the need for probabilistic methods of situating the Cy3 and Cy5 in close proximity to enable photoswitching. As proof of principle, these heterodimers have been applied to superresolution imaging of the tubular stalk structures of live Caulobacter crescentus bacterial cells (yellow in the figure).
Covalent heterodimers of the Cy3 and Cy5 fluorophores have been prepared from commercially available starting materials and characterized at the single-molecule level. This system behaves as a discrete molecular photoswitch, in which photoexcitation of the Cy5 results in fluorescence emission or, with a much lower probability, causes the Cy5 to enter into a long-lived, but metastable, dark state. Photoinduced recovery of the emissive Cy5 is achieved by very low intensity excitation (5 W/cm2) of the Cy3 fluorophore at a shorter wavelength. A similar system consisting of proximal, but not covalently linked, Cy3 and Cy5 has found application in stochastic optical reconstruction microscopy (STORM), a single-molecule localization-based technique for superresolution imaging that requires photoswitching. The covalent Cy3-Cy5 heterodimers described herein eliminate the need for probabilistic methods of situating the Cy3 and Cy5 in close proximity to enable photoswitching. As proof of principle, these heterodimers have been applied to superresolution imaging of the tubular stalk structures of live Caulobacter crescentus bacterial cells (yellow in the figure).
N. R. Conley, J. S. Biteen, and W. E. Moerner, J. Phys. Chem. B Letters 112, 11878 (2008) [![]() Slide] [journal link: JPC B Letter]
Slide] [journal link: JPC B Letter]
Single
Molecules of Bacterial Actin MreB Undergo Directed
Motion in Caulobacter Crescentus Cells4
S. Y. Kim, Z. Gitai, A. Kinkhabwala, L. Shapiro, and W. E. Moerner, PNAS 103, 10929 (2006) [![]() Full
Text] [Journal
link] [Supporting
material] [movie1]
[movie2]
[movie3]
[movie4]
Full
Text] [Journal
link] [Supporting
material] [movie1]
[movie2]
[movie3]
[movie4]
- E. Betzig, G.H. Patterson, R. Sougrat, O.W. Lindwasser, S. Olenych, J.S. Bonifacino, M.W. Davidson, J. Lippincott-Schwartz, and H.E. Hess, "Imaging intracellular fluorescent proteins at nanometer resolution," Science, 2006, 313, 1642-1645; M.J. Rust, M. Bates, and X. Zhuang, "Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM)," Nature Meth., 2006, 3, 793-795; S.T. Hess, T.P. Girirajan, and M.D. Mason, "Ultra-High Resolution Imaging by Fluorescence Photoactivation Localization Microscopy," Biophys. J., 2006, 91, 4258-4272. W. E. Moerner, “New Directions in Single-Molecule Imaging and Analysis,” Invited Perspective, Proc. Nat. Acad. Sci. (USA) 104, 12596-12602 (2007).
- S. W. Hell, "Far-Field Optical Nanoscopy," Science 2007, 316, 1153-1158.
- R.M. Dickson, A.B. Cubitt, R.Y. Tsien, and W.E. Moerner, "On/off blinking and switching behaviour of single molecules of green fluorescent protein," Nature, 1997, 388, 355-358.
- S.Y. Kim, Z. Gitai, A. Kinkhabwala, L. Shapiro, and W.E. Moerner, "Single molecules of the bacterial actin MreB undergo directed treadmilling motion in Caulobacter crescentus," PNAS, 2006, 103, 10929-10934.